Definition of Homology, Orthology and Paralogy
- Homology is a term used when two genes share a common ancestor. The two genes are considered “homologous” or “not homologous” depending on if they are related or not related. It is a common mistake to say “slightly or significantly homologous” and the term homologous should not be a substitute for similarity. When two sequences are similar, we can say that they show homology, they are homologues, or they are homologous. Homology can’t be confirmed since it is not possible to see into the past.
- Orthology is a type of homology that describes two genes that descended from a common ancestor divided by speciation. If two genes have a common ancestor and are found in two descent strains (genomes), we can say that they show orthology, they are orthologs, or they are orthologous.
- Paralogy, another type of homology, describes two genes divided by duplication within the genome. If two genes have a common ancestor and are found in the same strain (genome), we can say that they have paralogy, they are paraologues, or they are paralogous. There are many paralogues in bacterial genomes as most of the bacterial genes were created by duplication.
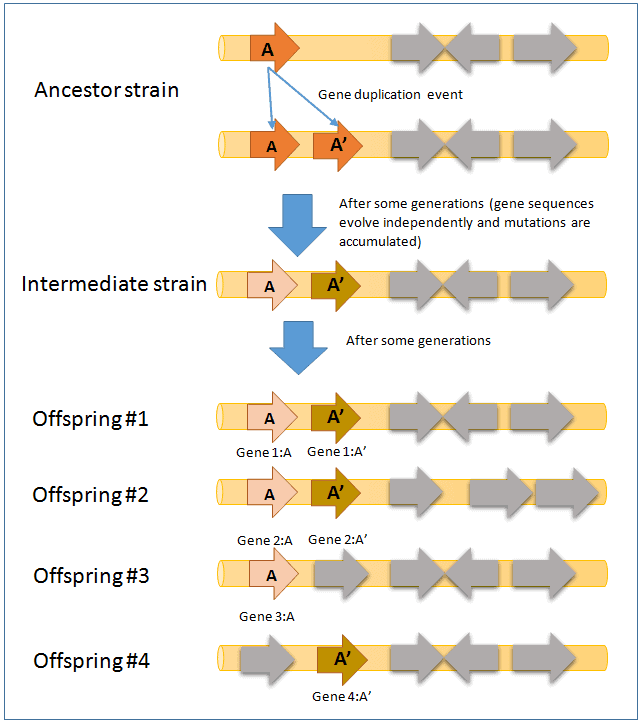
In the above figure, gene A in the ancestral strain was duplicated to give birth to gene A’. Over time, these two genes evolve independently and results in different sequences. As more time passes, the sequences become more different and as a result, genes A and A’ in the intermediate strain show paralogy.
The intermediate strain is the shared common ancestor to offsprings #1,2,3 and 4. After a considerable amount of time, gene A and A’ now confer different function when compared to the intermediate strain (e.g. gene A is involved in glucose utilization whereas gene A’ is involved in galactose utilization).
The following statements are true:
- All genes (Gene 1:A, 1:A’, 2:A, 2:A’,3:A, 4:A’) show homology. They will show significant similarity/identity in sequence comparison (by BLAST or uBLAST programs).
- Genes 1:A, 2:A and 3:A show orthology.
- Genes 1:A’, 2:A’ and 4:A’ show orthology.
- These pairs show paralogy (1:A and 1:A’, 2:A and 2:A’).
- In offspring #3 and #4, one of the genes is missing by a gene deletion event. Gene 3:A and 4:A’ show homology, but not orthology.
How to detect orthologs for large scale comparative genomics
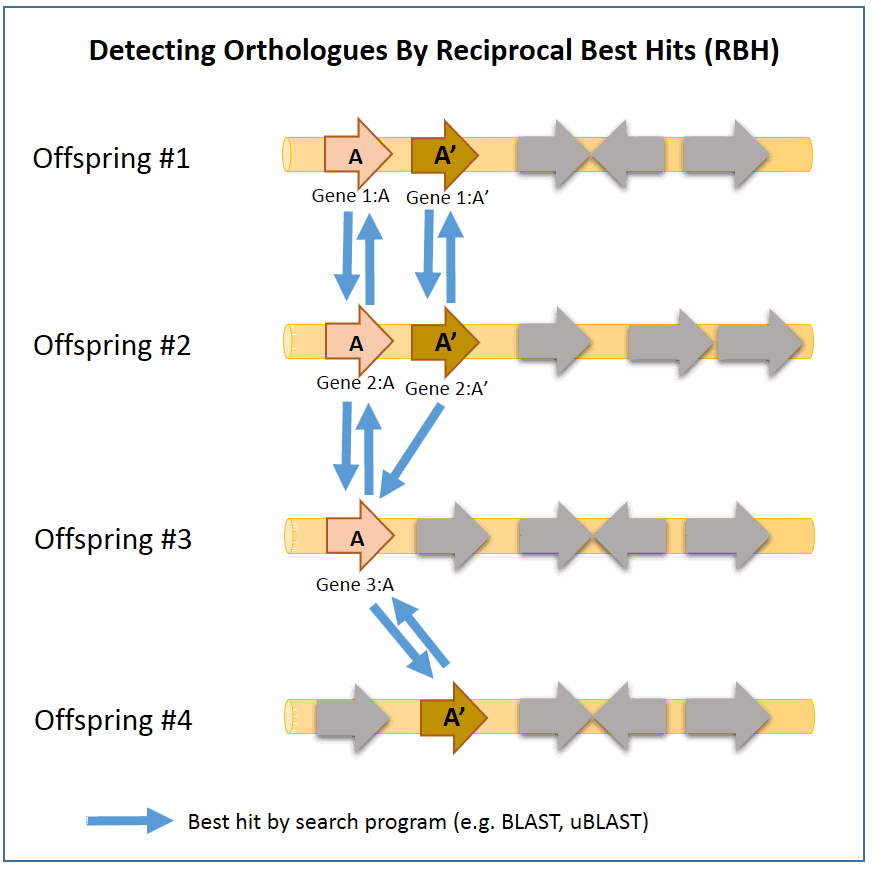
Usually, finding orthologs can be achieved using Reciprocal Best Hit (RBH) or ORF-independent methods.
1. Reciprocal Best Hit (RBH) method

In RBH method, two genomes are compared in a pairwise manner using amino acid sequences of each CDSs.
In the above example, orthologs can be confirmed by the homology search process as they will result as “best hits” to each other. Here, genes 2:A and 3:A are orthologs as they are “best hits” to each other. In contrast, genes 2:A’ and 3:A does not show orthology as only 2:A’ -> 3:A has the best-hit relationship (not reciprocal). In rare cases, using this method can give false results unfortunately. Here, genes 3:A and 4:A’ are detected as “orthologs”, which is not true. Orthology can be further investigated using a method called “tree reconciliation”, which will reveal that genes 3:A and 4:A’ are not really orthologous.
RBH is a commonly used method for large scale computation as it is suitable for such computation. RBH analysis can be carried out using BLAST or uBLAST program. In EzBioCloud Database, we use uBLAST with an e-value threshold of 10-6 (Ward & Moreno-Hagelsieb, 2014).
2. Open reading frame (ORF)-independent method
This method provides additional information and complements the RBH method mentioned above. It was used in a large scale comparative genomics study (Chun et al., 2009) in which basics of the algorithm were given. It is different from RBH method because:
- It uses nucleotide sequences of CDSs instead of amino acid sequences.
- Nucleotide sequences of CDSs of a query genome are searched against contig/chromosome sequences in subject genome. Therefore, it does not rely on gene prediction process (=defining ORF or CDS on contig sequences).
- RBH method identifies orthologs, whereas (ORF)-independent method finds orthologous DNA regions.
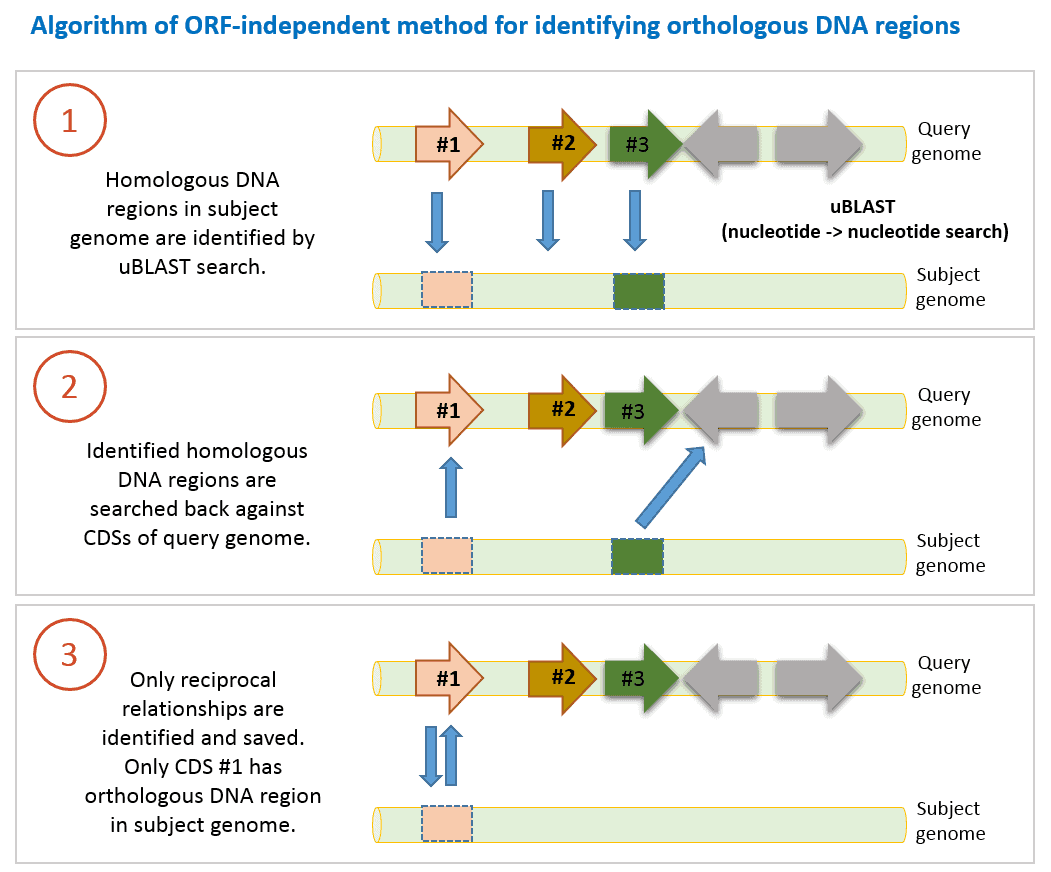
The algorithm used in EzBioCloud Database are as follows:
- DNA sequence of a CDS in query genome is searched against contig sequences in a subject genome using uBLAST program with an e-value threshold of 10-6. This nucleotide-nucleotide comparison can identify homologous DNA region in the subject genome. We only take it into account if query coverage is >70% (at least 70% of query DNA sequence of a CDS is matched to the hit region in the subject genome).
- Identified homologous DNA region (we do not know if it is orthologous yet) is searched back with uBLAST against DNA sequences of all CDSs of query genome.
- If search results are reciprocally hit DNA region is now called orthologous DNA region.
- 1-3 steps are carried out for all CDSs of query genome.
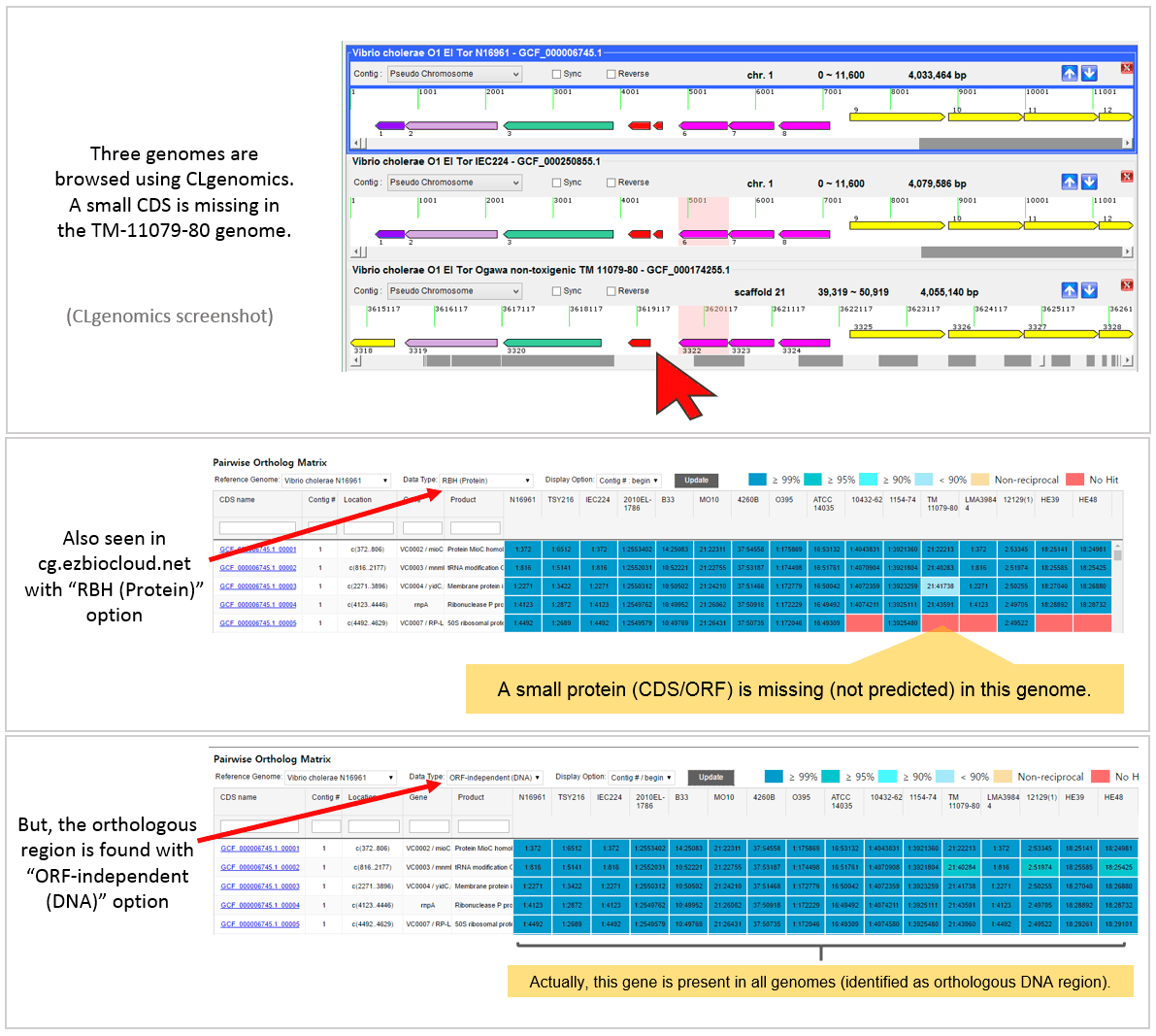
The below example highlights how ORF-independent method helps ortholog identification when ORF/CDS prediction is not correct.
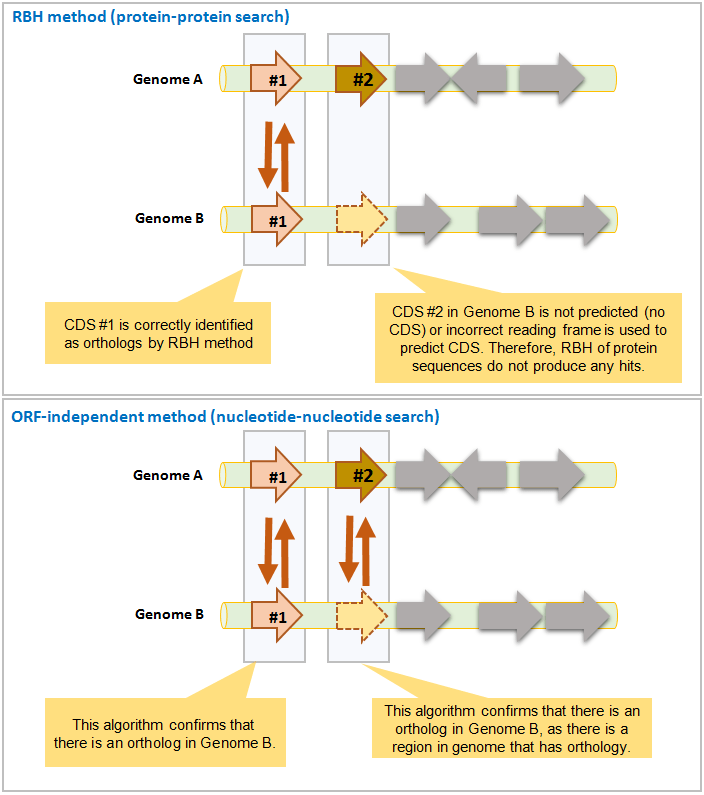
A real example is given below. This is taken from “Vibrio cholerae tutorial set” available at https://ezbiocloud.net/genome/view_myCGData where CLG files for all genomes can be obtained. CLgenomics should be installed to open and browse CLG files. The program is freely available at https://data.cjbioscience.com/software/clgenomics.
Pan-genome Orthologous Groups (POGs)
Orthologs in multiple genomes can be clustered to generate non-redundant “orthologous groups”. Orthologous groups then forms the pan-genome of all genomes included in the pan-genome analysis, so it is called “Pan-genome Orthologous Groups (POGs)”.
Learn more about Pan-genome Orthologous Groups (POGs)
References
- Chun, J. et al. Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proc Natl Acad Sci U S A 106, 15442-15447 (2009).
- Ward, N. & Moreno-Hagelsieb, G. Quickly finding orthologs as reciprocal best hits with BLAT, LAST, and UBLAST: how much do we miss? PLoS One 9, e101850 (2014).
Last updated on May 10th, 2016 (EK)

