
In the recent paper (Kim et al., 2015), we evaluated DNA G+C content data obtained by HPLC and thermal melting experiments (Tm) with more accurate experiments directly calculated from whole genome sequences. The range of DNA G+C ratio is different by species, making it a unique characteristic for each species. For example, more variations in DNA G+C content are found among species found in nature (e.g Psudomonas) versus species that are pathogenic to humans (e.g. Salmonella). In any case, the range of DNA G+C ratio among species (as defined by Average Nucleotide Identity) is 1%. However, experimentally determined DNA G+C ratio values, reported in past publications, showed high levels of errors when compared with genome-derived values. In fact, about half of these values showed over a 1% difference. The DNA G+C ratio can only be meaningful when the errors are reasonably small. I believe that these levels of errors are not useful and should be rectified using genome sequence data. In the current NGS era, why bother publishing values that are soon to be correctly (likely within a year or two)?
The following figure shows how DNA G+C ratios vary among species, but mostly within a 1% range.
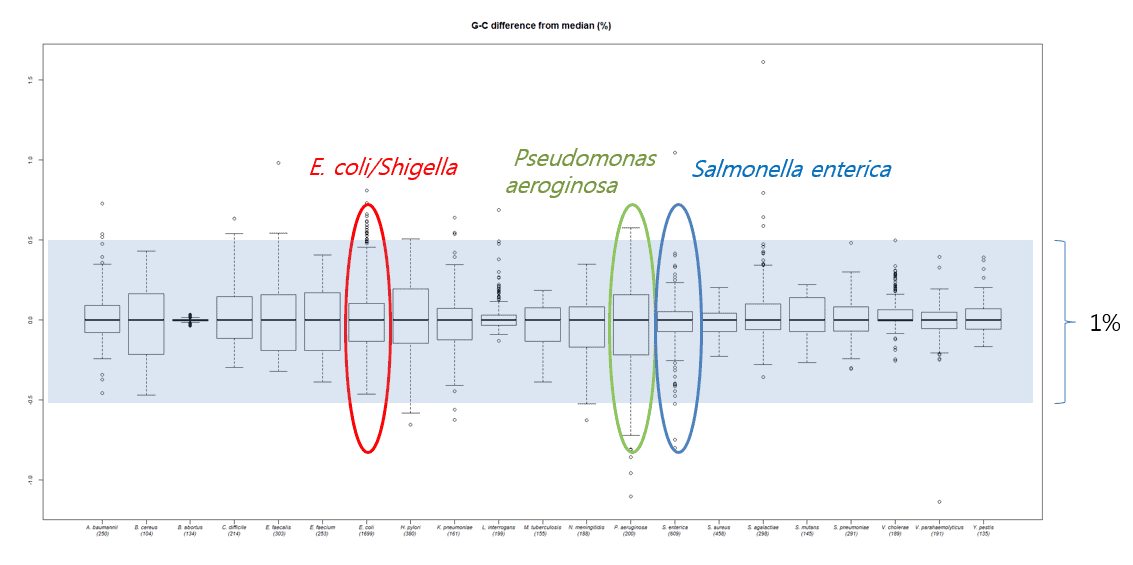
1. Kim, M., Park, S.C., Baek, I. & Chun, J. Large-scale evaluation of experimentally determined DNA G+C contents with whole genome sequences of prokaryotes. Syst Appl Microbiol 38, 79-83 (2015).
By Jon Jongsik Chun (CEO of CJ Bioscience, Inc. & Professor at Seoul National Univ.)
Updated on April 4th 2016

