이 Tutorial은 건강한 사람과 환자의 호흡기 샘플을 비교하여 분석하는 과정을 소개하는 설명서입니다. 내용을 따라가면 EzBioCloud의 microbiome taxonomic profiling(MTP) 사용법을 손쉽게 배울 수 있습니다.
Tutorial에 사용된 데이터는 2014년 Hana Yi et al 에 의해 발표된 자료이며, 이는 EzBioCloud 데이터베이스에도 실려 있습니다. 데이터를 사용자 계정에 추가하는 것부터 시작하겠습니다.
www.EzBioCloud.com에 로그인 하기
천랩 홈페이지나 EzBioCloud에 가입된 계정으로 로그인할 수 있습니다. 만약 계정이 없다면 먼저 가입을 진행해야 합니다.
Microbiome Taxonomic Profiling (MTP) 서비스 페이지로 이동하기
EzBioCloud의 메인 페이지에서 [Apps] -> Microbiome Taxonomic Profiling (MTP)를 차례로 클릭합니다.
[EzBioCloud DB] 열기
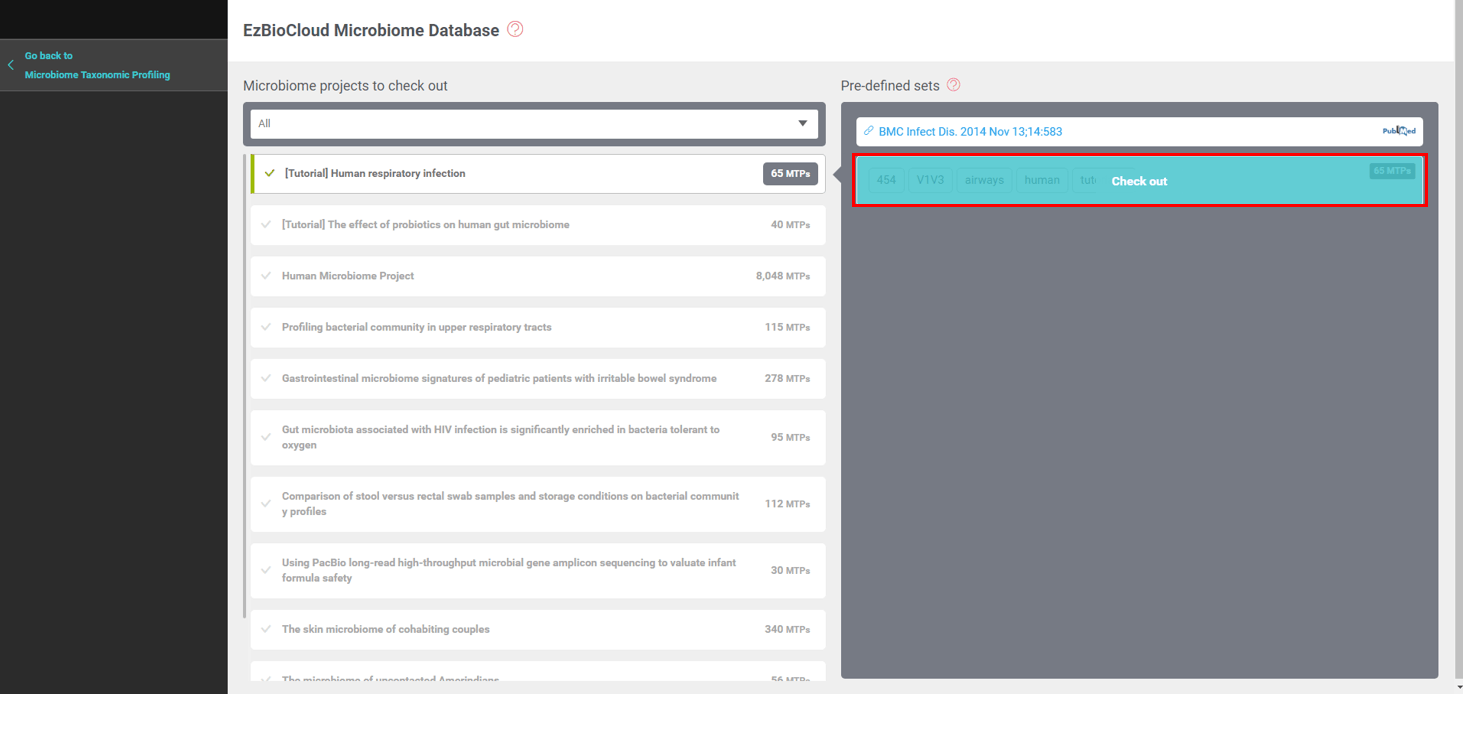
EzBioCloud DB는 분석에 활용할 수 있는 많은 데이터 세트를 보유하고 있습니다. 지속적으로 업데이트되고 있는 데이터베이스이며, 여기에는 Human Microbiome Project (HMP)가 사람 신체 부위 19곳에서 얻은 8,048개의 MTP 결과도 포함되어 있습니다.먼저 EzBioCloud DB에서 미리 구성된 “[Tutorial] Human respiratory infection” 데이터 세트를 선택하여 추가합니다. 이 데이터 세트는 65개의 MTP로 구성되어 있습니다.
계정에 보유한 MTP 확인하기
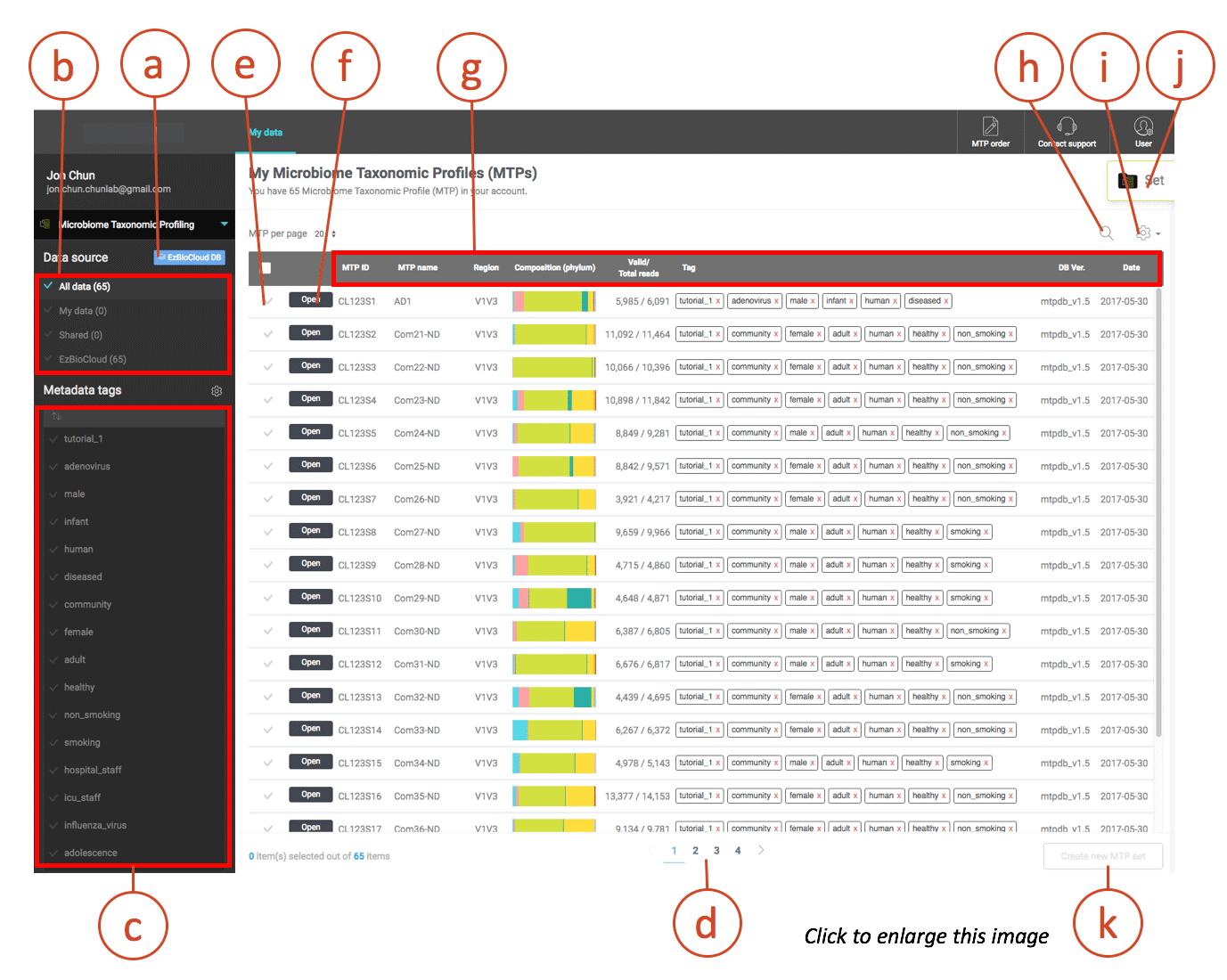
Tutorial 세트를 추가했다면 metadata tag가 달린 65개의 MTP가 생겼을 것입니다. Metadata tag는 많은 수의 마이크로바이옴 샘플을 다룰 때 유용하게 사용할 수 있는 기능입니다. 데이터를 구조화하거나 바이오마커, 유의미한 taxa를 찾을 때 중요한 역할을 할 것입니다. 아래의 스크린샷을 봅시다.
Public database의 마이크로바이옴 데이터베이스는 수많은 MTP를 가지고 있으며, 사용자가 자유롭게 이들을 분석에 활용할 수 있습니다. 천랩의 목표는 사용자 친화적인 EzBioCloud의 인터페이스를 통해 표준화된 마이크로바이옴 데이터 세트를 제공하는 것입니다.
사용자는 다양한 라이브러리에서 MTP를 선택하여 보유할 수 있습니다.
[MyData] – 천랩에 의해 분석된 나의 데이터입니다.
[Shared] – 다른 연구자가 공유한 데이터입니다.
[EzBioCloud] – EzBioCloud DB에서 선택한 데이터입니다.
사용자의 계정에 있는 metadata tag 리스트입니다. 태그를 클릭하면 쉽고 빠르게 MTP를 선택할 수 있으며, 이를 이용하여 MTP set을 생성한 후 더 깊이 있는 분석을 수행할 수 있습니다.
이 예제에 포함된 MTP 65개는 한 페이지에 다 보이지 않으므로 다른 샘플을 살펴보려면 페이지를 이동하여야 합니다.
체크 표시를 클릭하여 각 MTP를 선택하거나 선택 해제할 수 있습니다.
[Open]을 클릭하면 해당하는 MTP에 대해 “Single MTP Browser”가 실행됩니다.
MTP에서 사용되는 용어는 아래를 참고 바랍니다.
“MTP name”은 천랩 또는 사용자가 샘플(데이터 포함)에 부여한 이름입니다.
“Region”은 유전자의 시퀀싱 영역입니다(e.g.V1 to V9 for 16S). 이 정보는 “Toggle columns (i)”로 숨길 수 있습니다.
Phylum 수준의 구성을 표시하여 빠르게 비교해 볼 수 있도록 하였습니다.
이 컬럼은 “Valid/Total reads”를 표시합니다.
“(Metadata) Tags”는 샘플(MTP)을 가장 잘 설명하는 간단한 단어입니다. 태그를 조합하여 여러 개의 MTP set을 구성하고 분류할 수 있습니다.
“Date”는 천랩의 파이프라인을 통해 MTP가 생성된 날짜입니다. (샘플 채취 날짜와는 다릅니다.)
계정에 포함된 MTP를 검색할 수 있습니다.
개별 컬럼을 숨기거나 나타나게 세팅을 변경할 수 있습니다.
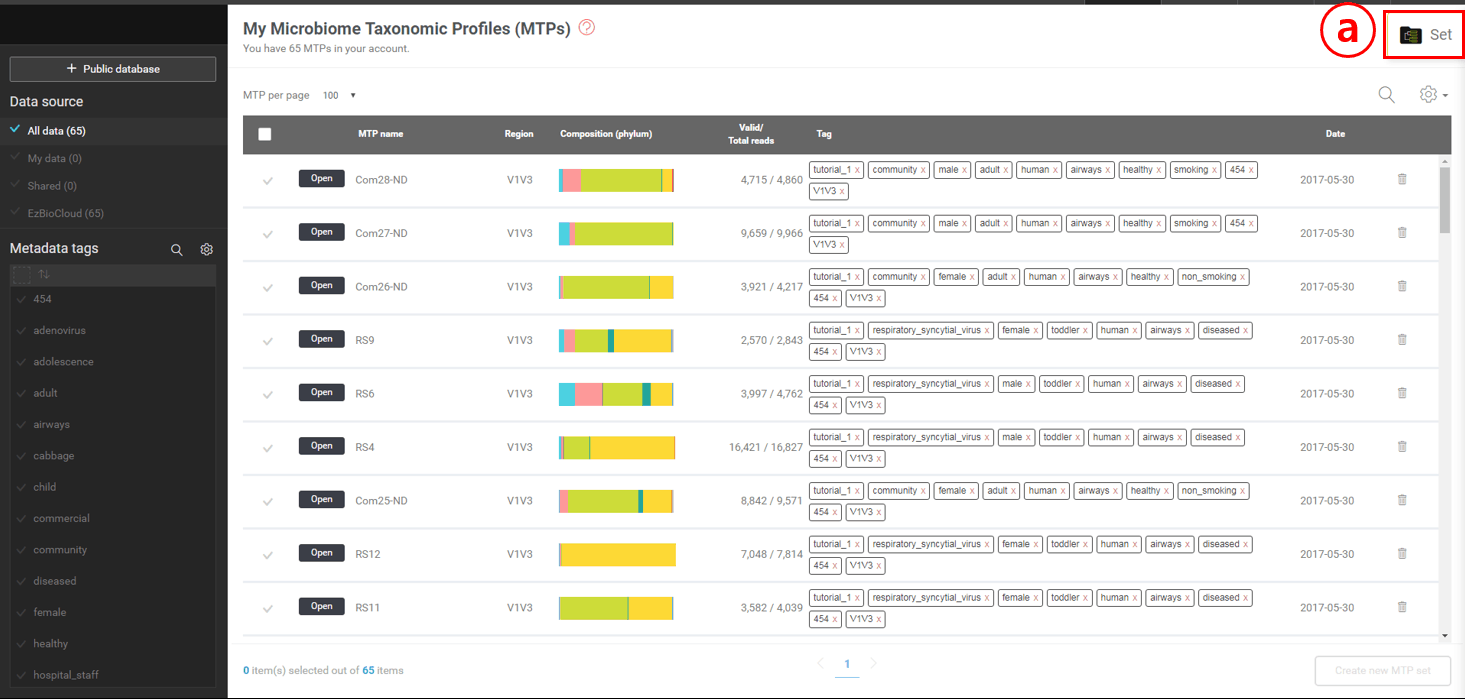
하나 이상의 MTP set을 만들고 싶다면 “MTP set”으로 이동합니다.
여러 개의 MTP를 선택한 후 MTP set을 생성합니다.
“Single MTP Browser”를 이용하여 개별 MTP 살펴보기
앞선 과정을 통해 호흡기에서 채취한 샘플의 MTP 65개를 가지게 되었습니다. 미생물 군집 구조의 분석은 Roche 454 플랫폼으로 16S V1-V3 영역을 시퀀싱하여 진행되었습니다. EzBioCloud 데이터베이스를 이용하므로 각 시퀀싱 리드를 종 수준으로 동정하는 것이 가능합니다. 각각의 MTP는 샘플의 미생물 군집 구조에 대해 완벽한 정보를 담고 있습니다.
AD1 (MTP ID=CL123S1)라는 MTP샘플을 [Open]을 클릭하여 불러옵니다. 태그에 의하면 이 샘플은 아데노바이러스에 감염된 남성 유아로부터 채취한 호흡기 샘플입니다. 이 샘플에 대해 탐색할 수 있는 새로운 웹 페이지(탭)가 열릴 것입니다.
이 MTP로 “About MTP” 탭에서 AD1 샘플에 대한 다음과 같은 정보를 찾을 수 있습니다.
퀄리티 필터 작업 후의 총 리드 수는 6,091개입니다.
이 중 18개의 리드는 16S rRNA 유전자가 아닌 것으로(non-specific) 판명되었습니다.
88개의 키메라 리드가 발견되어 제거되었습니다.
그러므로 유효 리드 수는 5,985개이며 평균 리드 길이는 481.6 bp입니다.
유효 리드의 92.5% (5.539개 리드)는 EzBioCloud DB와 천랩의 파이프라인에 의해 종 수준으로 동정 되었으며, 총 83개의 종이 이 샘플에서 발견되었습니다.
“Alpha diversity” 탭을 누르면 종 풍부도(species richness, 샘플의 미생물 종의 수를 추산한 값)와 종 균등도(species evenness, or diversity index)를 볼 수 있습니다.
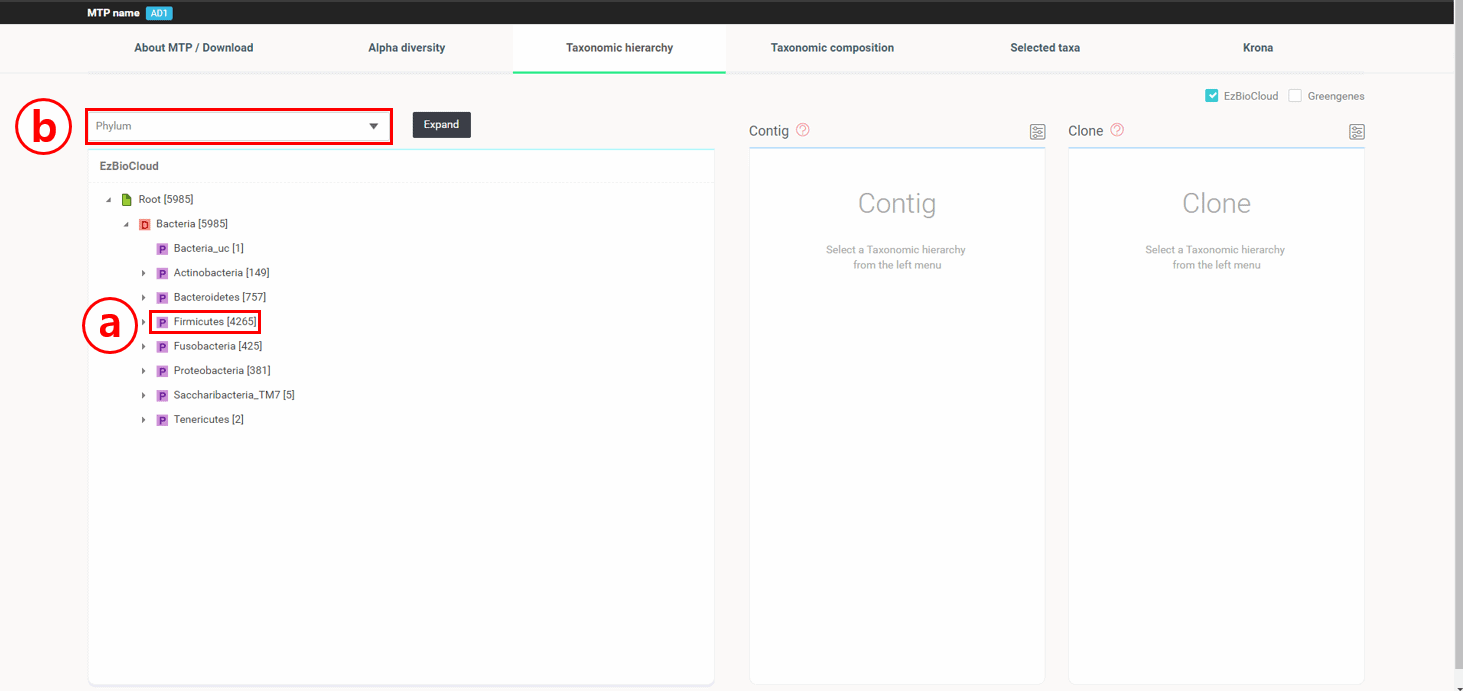
“Taxonomic Hierarchy”탭을 선택하면 16개 read가 taxonomic hierarchy 구조에 따라 표시됩니다.
4,265 리드가 Firmicutes phyum에 속합니다. Taxon 이름 왼쪽의 회색 삼각형을 클릭하여 트리를 펼칠 수 있습니다.
또 다른 방법으로는 “Species”를 선택하고 [Expand] 버튼을 눌러 모든 트리 하위 구조가 펼쳐지게 할 수 있습니다.
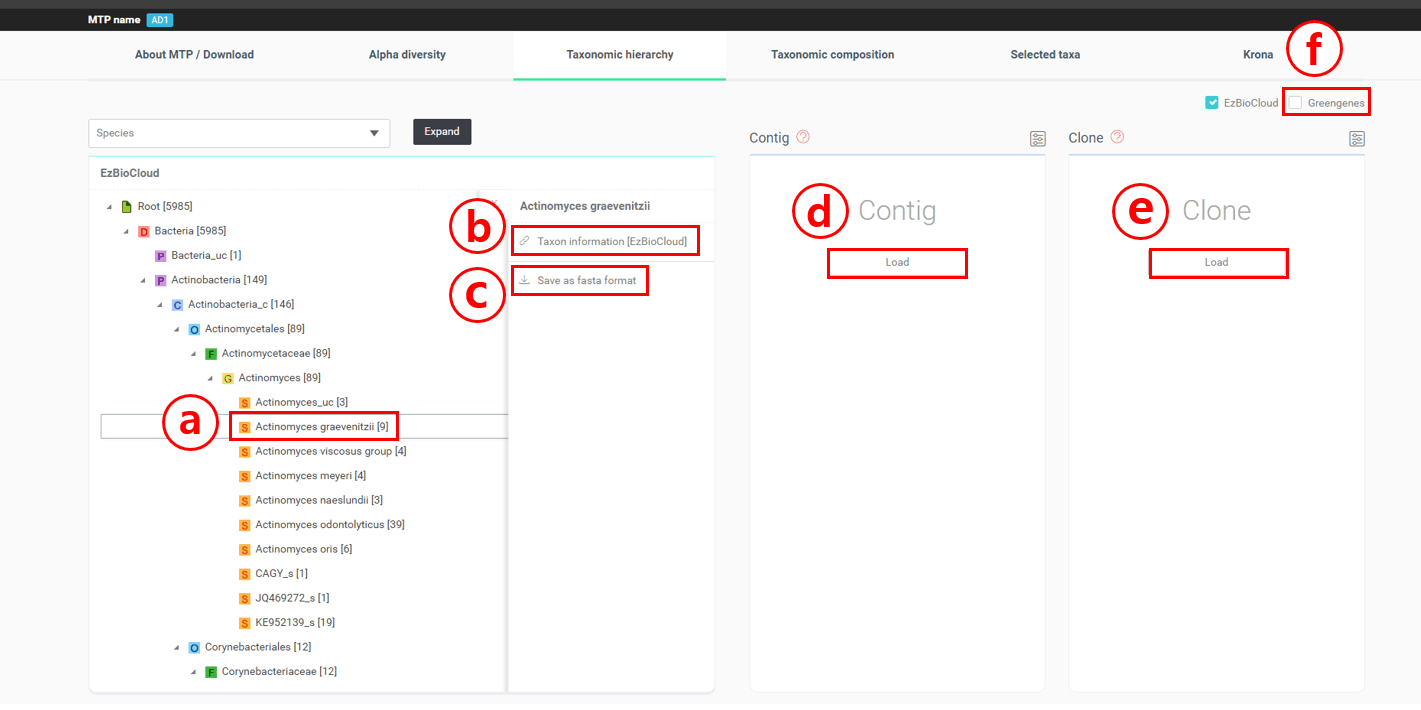
Taxonomic hierarchy를 완전히 펼쳤다면, 이제 Actinomyces graevenitzii를 자세히 살펴봅시다. Actinobacteria (phylum); Actinobacteria_c (class); Actinomycetales (order); Actinomycetaceae (family); Actinomyces (genus)의 하위에 있는 “Actinomyces graevenitzii”를 클릭합니다.
Actinomyces graeventitzii를 클릭하여 필수 메뉴를 가져옵니다.
Taxon 정보를 EzBioCloud 데이터베이스에서 불러오려면 이곳을 클릭합니다. Actinomyces graeventitzii의 taxon 정보를 가지고 한 번 살펴보겠습니다. EzBioCloud 데이터베이스는 taxon에 대한 다양한 분류학적, 생물학적 정보를 제공할 뿐만 아니라 건강한 사람의 신체에 이 종이 얼마나 분포해 있는지도 알려줍니다. 하단의 차트를 보면 Actinomyces graeventitzii은 사람의 구강에 약 1%정도 서식한다는 것을 알 수 있습니다(see here).
Actinomyces graeventitzii로 동정된 모든 시퀀스를 FASTA file로 다운로드합니다.
[Load]를 눌러 Actinomyces graeventitzii로 동정된 컨티그(contigs)를 불러옵니다. 컨티그는 같은 template에서 유래하여 동일한 것으로 간주되는 시퀀스의 그룹을 말합니다.
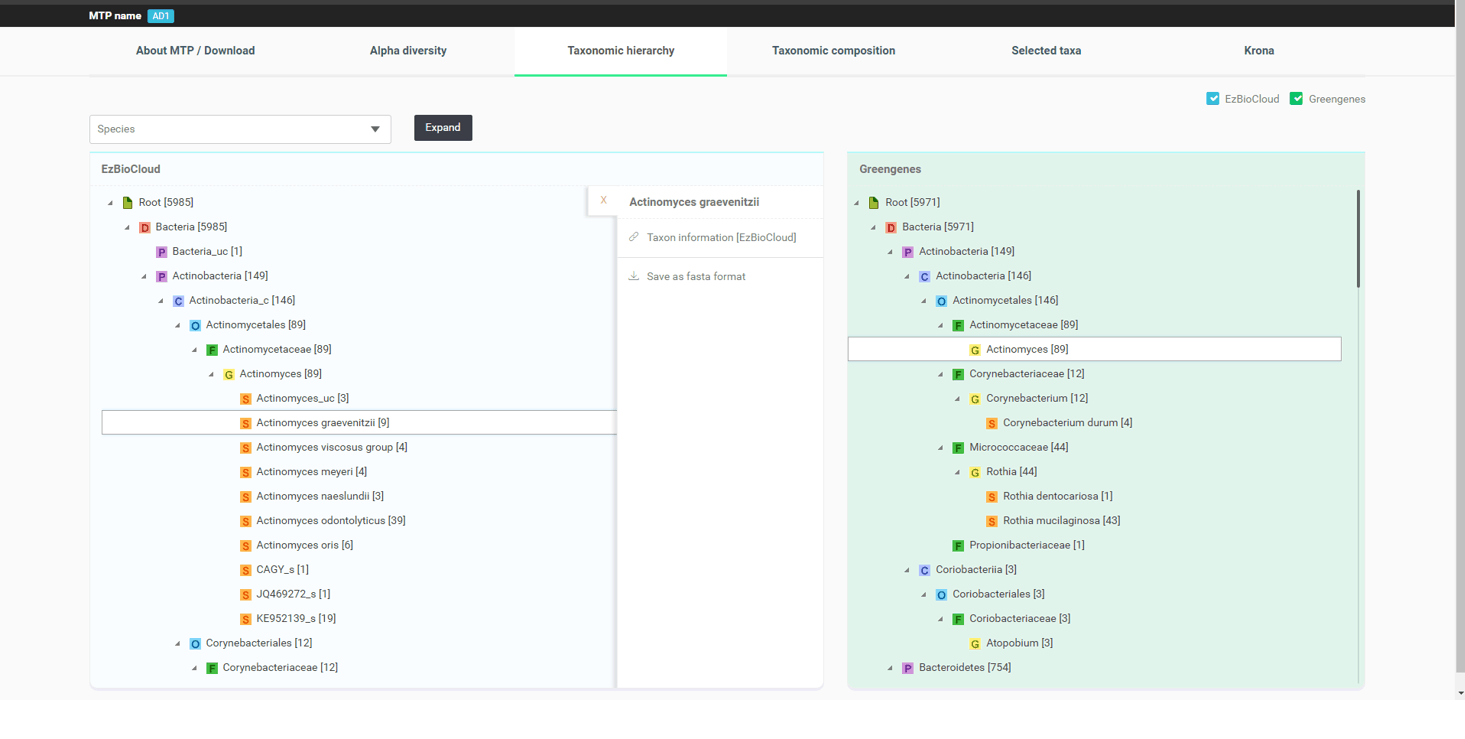
“QIIME/Greengenes”라는 박스를 체크하면 동일한 샘플에 대한 analogous taxonomic hierarchy가 표시됩니다. 이것은 Greengenes 데이터베이스와 QIIME 파이프라인을 통해 분석된 것으로, 두 분석 결과를 함께 보면서 비교할 수 있습니다. 아래의 이미지는 genus Actinomyces와 genus Corynebacterium의 분석 결과의 차이를 보여주고 있습니다.
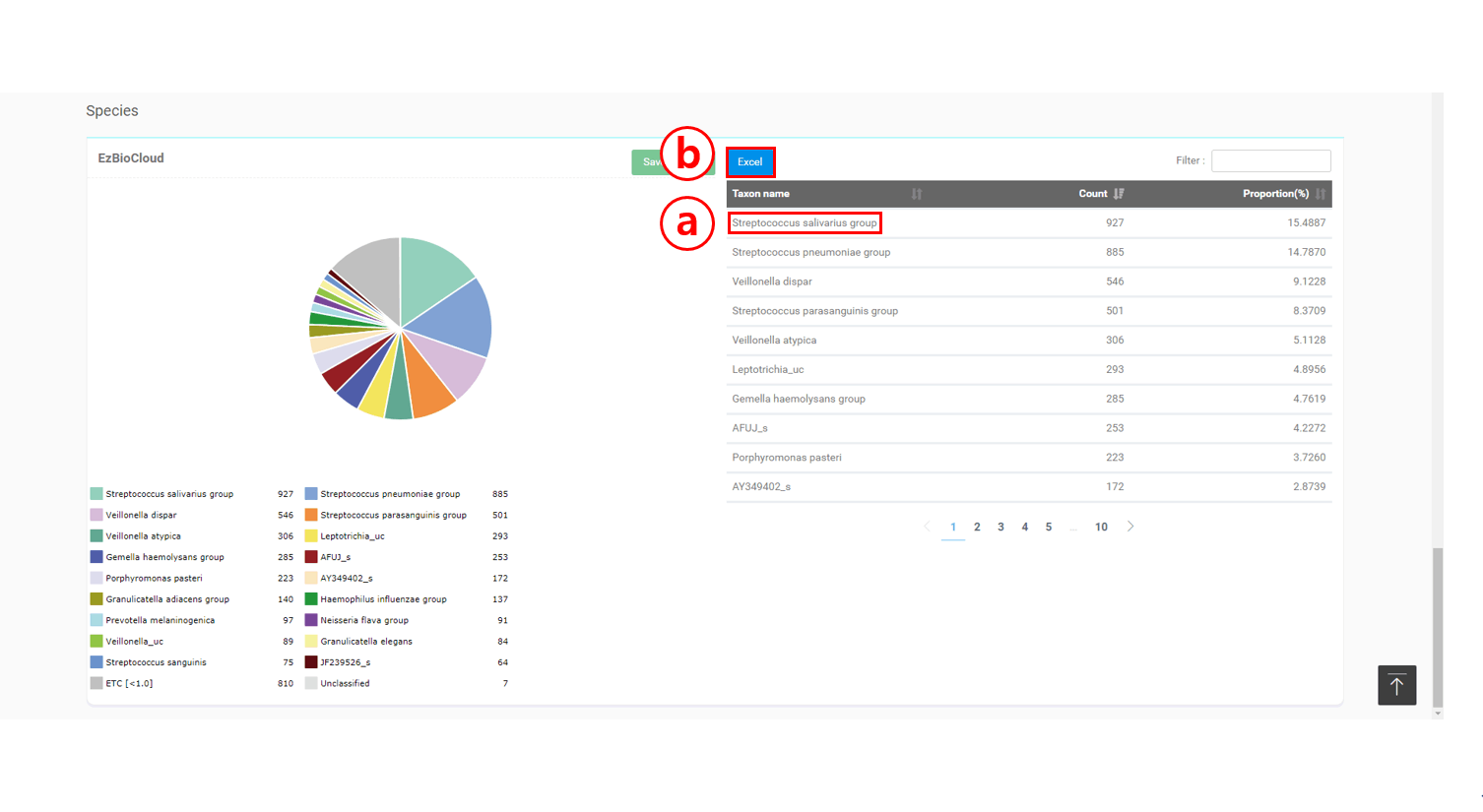
“Taxonomic composition” 탭에서 모든 taxonomic rank (phylum to species)의 양적인 구성은 테이블과 파이 차트로 주어집니다. 직접 데이터를 다루고 싶은 분들을 위해 Excel 형식으로 데이터를 다운로드하는 옵션도 가지고 있습니다. AD1 샘플에서 가장 풍부한 종은 “Streptococcus salivarius group” (15.5%)이며, “Streptococcus pneumoniae group” (14.8%)과 Veillonella dispar (9.1%)가 그 뒤를 따르고 있습니다. “taxonomic group”은 16S rRNA 유전자만으로는 구분되지 않는 여러 개의 종을 포함하고 있는 그룹입니다. “Streptococcus salivarius group”을 클릭해서 이 그룹을 구성하는 종들을 설명하는 웹 페이지로 이동할 수 있습니다
클릭하면 Taxon을 설명하는 웹 페이지로 이동합니다.
클릭하면 데이터를 Excel 파일로 저장합니다.
Krona 탭에서는 Krona tool로 taxonomic composition 정보를 불러온 결과를 보여줍니다. Krona tool은 오픈소스 시각화 프로젝트이며 여기에서 이용 가능합니다.
MTP 데이터라면 어떤 것이든 지금까지 설명한 방법으로 탐색할 수 있습니다.
MTP set 생성하기
마이크로바이옴 연구의 주요한 목적 중 하나는 샘플 여러 개를 그룹으로 묶어서 분류학적인 특징을 파악하는 것입니다. MTP set은 EzBioCloud에서 MTP의 묶음으로 정의됩니다. 이용자는 직접 샘플을 선택해서 set을 만들거나, (metadata) 태그를 이용하여 어느 정도 자동적으로 생성할 수 있습니다.
Tutorial set에서는 태그를 다양하게 조합하여 set을 생성할 것입니다. “Healthy”와 “Diseased”로 정의되는 두 개의 set을 각각 만들어 봅시다.
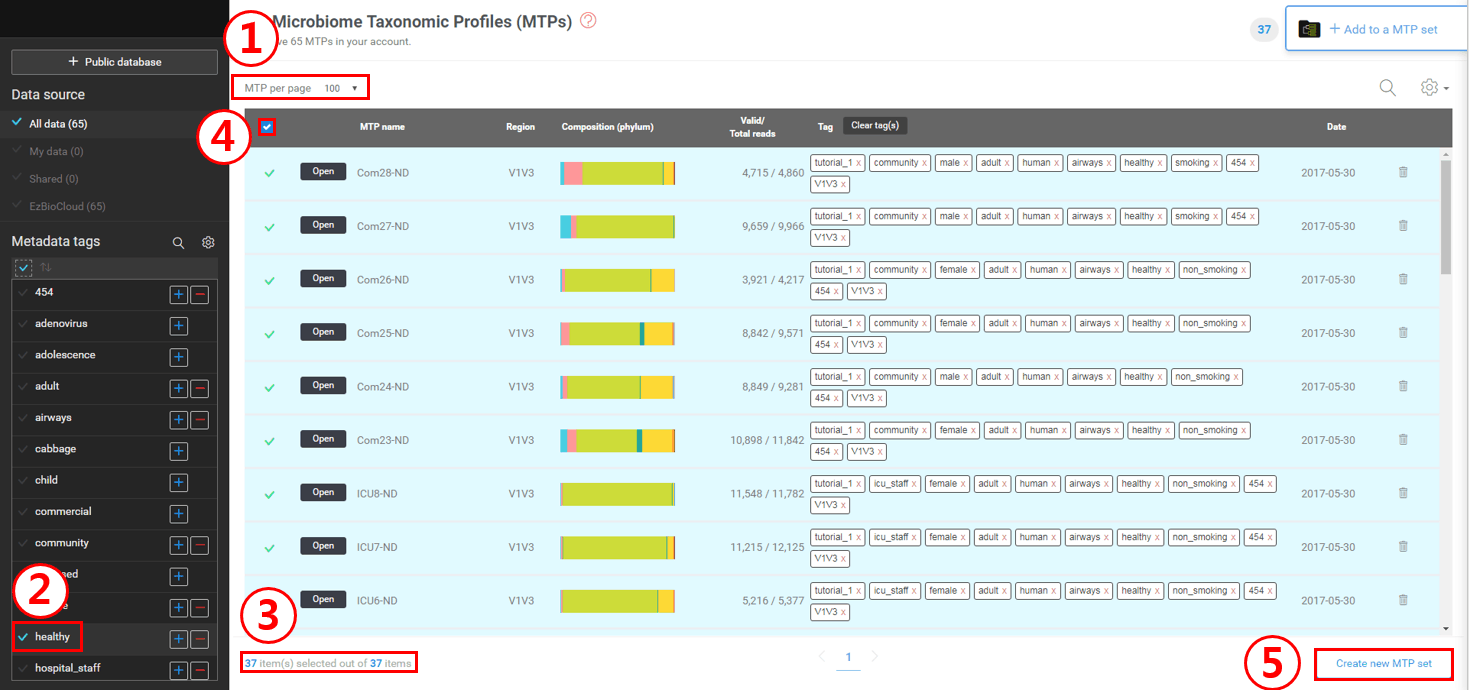
첫째로, “MTP per page”를 한 페이지에 100개까지 볼 수 있도록 설정합니다.
태그 패널(왼쪽)에서 “healthy” 태그를 체크하여 이 태그가 달린 MTP만 선택합니다..
“healthy” 태그가 달린 37개의 MTP만 리스트에 나타날 것입니다.
이 박스를 체크하여 모든 37개의 MTP를 선택합니다.
[Create new MTP set]을 클릭하여 MTP set을 생성한 후 “Healthy”라고 이름을 붙입니다.
똑같은 방법으로 “Diseased” 태그를 이용하여 MTP set을 만드십시오. 이 셋은 28개의 MTP를 포함할 것입니다(아래 참조).
“Diseased” set은 28개의 MTP를 가지고 있습니다.
“Healthy” set은 37개의 MTP를 가지고 있습니다.
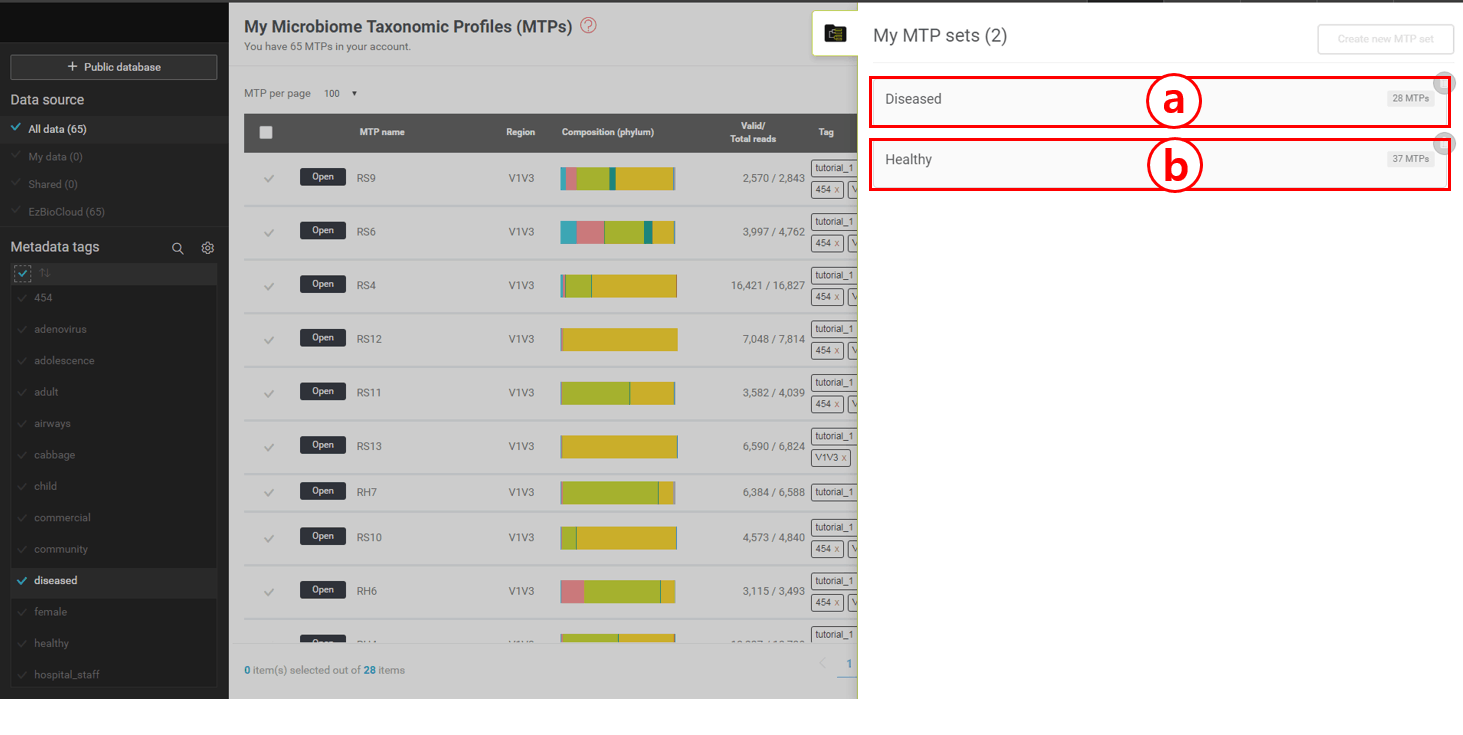
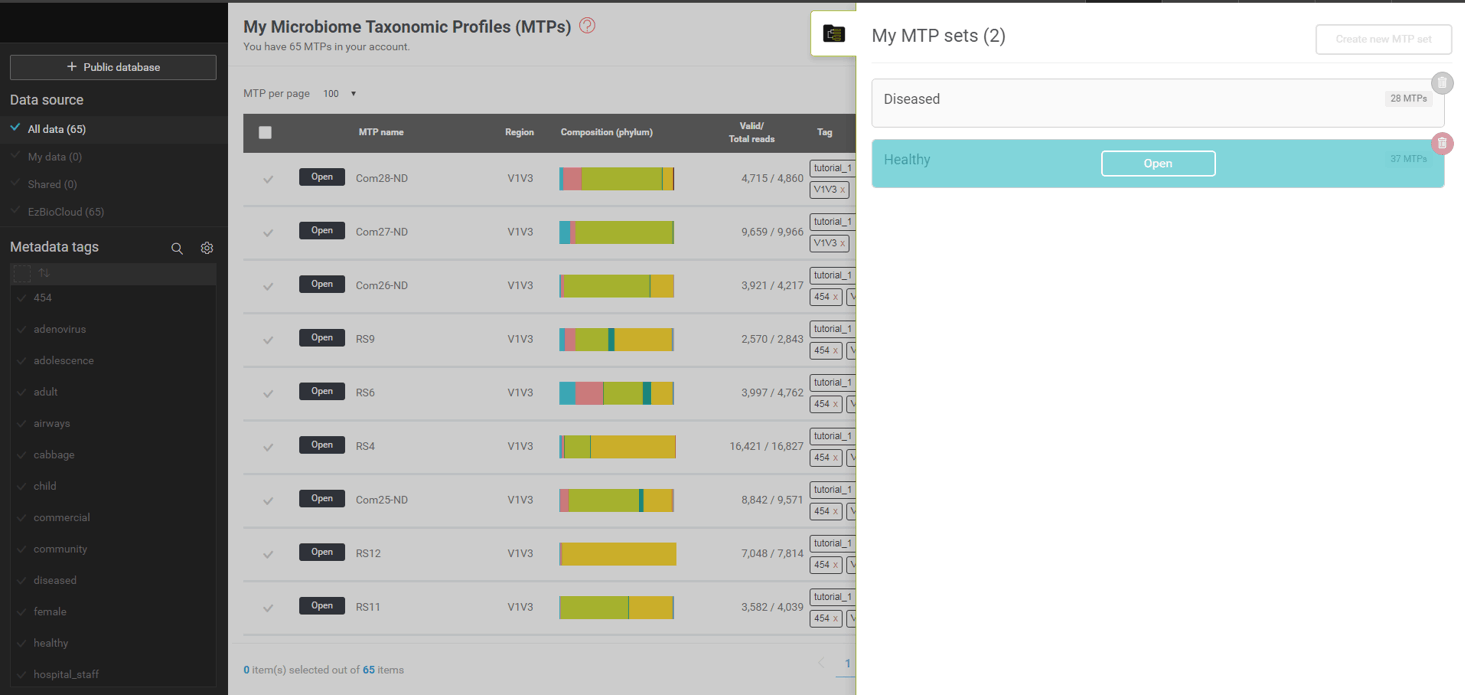
MTP set의 마이크로바이옴 정보를 살펴보기 위해 set을 나타내고 있는 박스(위 스크린샷의 a와 b) 위에 마우스 커서를 이동합니다.
이 탭을 클릭하여 MTP set을 선택하는 메뉴를 불러올 수 있습니다.
MTP set 열기
하나의 MTP set은 여러 개의 MTP로 이루어져 있으며 많은 통계적 분석의 처리 단위로 간주됩니다. 우리는 “healthy”와 “diseased” 두 개의 MTP set을 만들었습니다. 첫째로 열어 볼 것은 “healthy” set입니다. 이 set를 구성하는 MTP는 건강한 사람 37명의 호흡기에서 채취한 샘플로 16S에 기반한 미생물 군집 분석을 통해 만들어졌습니다. MTP set을 불러온 후 (앞의 스크린샷의 (a)) “healthy” set에 커서를 올리면 [Open] 버튼이 나타날 것입니다. 이 버튼을 누르면 “MTP set browser”를 열 수 있습니다. (아래 스크린샷 참조)
“Healthy” set을 보여주고 있는 MTP set browser (아래)
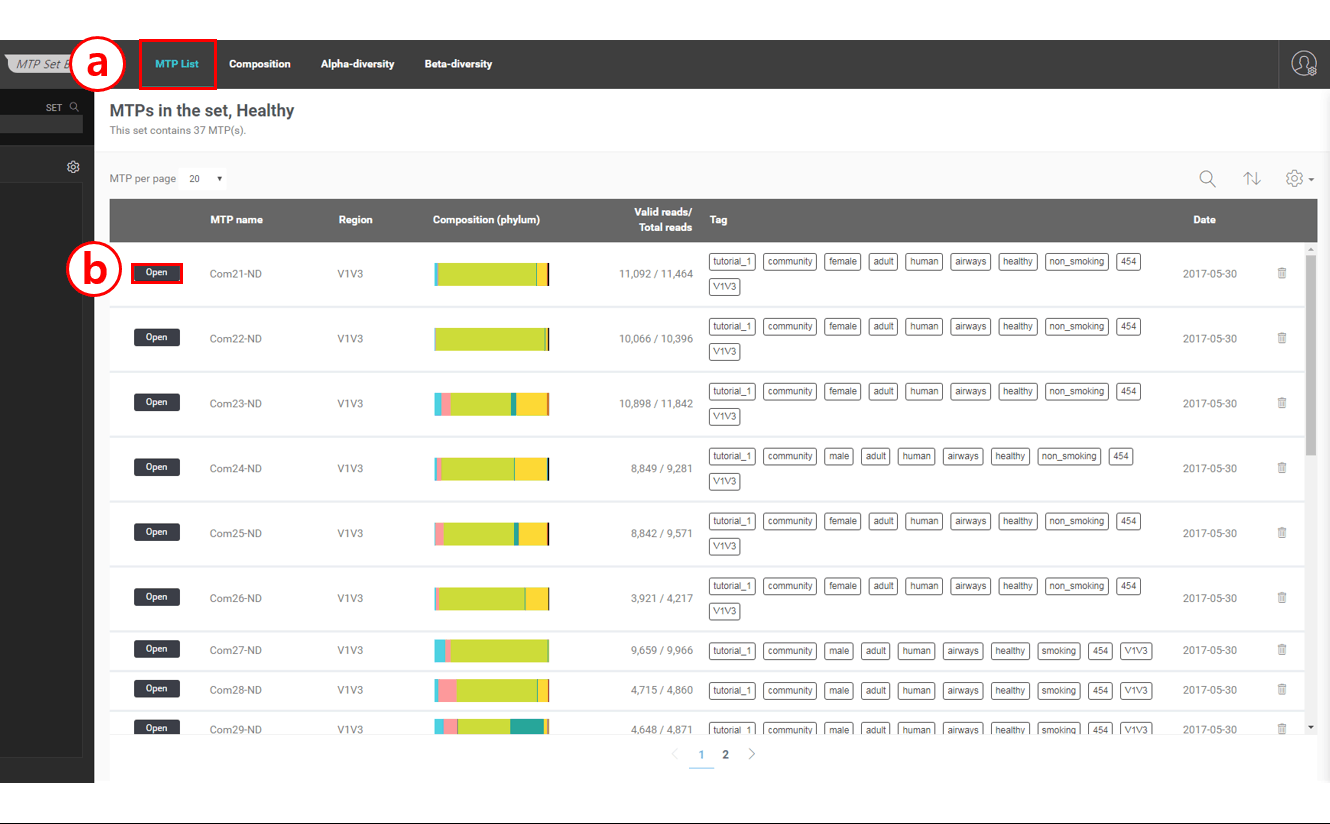
주 메뉴
[Open]을 클릭하면 Single MTP Browser로 각 MTP의 데이터를 탐색할 수 있습니다.
주 메뉴에는 4개의 항목이 있습니다.
MTP List: Set에 포함된 MTP 목록을 보여줍니다.
Composition: Set의 taxonomic composition 통계 자료를 제공합니다.
Alpha-diversity: Set의 다양성 지수에 대한 통계적 요약을 제공합니다.
Beta-diversity: 베타 다양성 분석(MTP 사이의 상관관계 분석) 결과를 제공합니다.
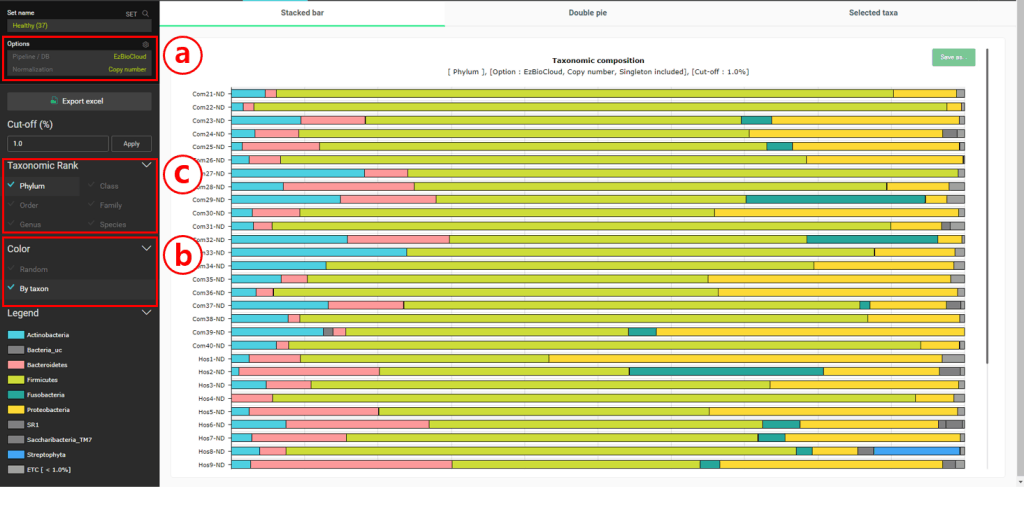
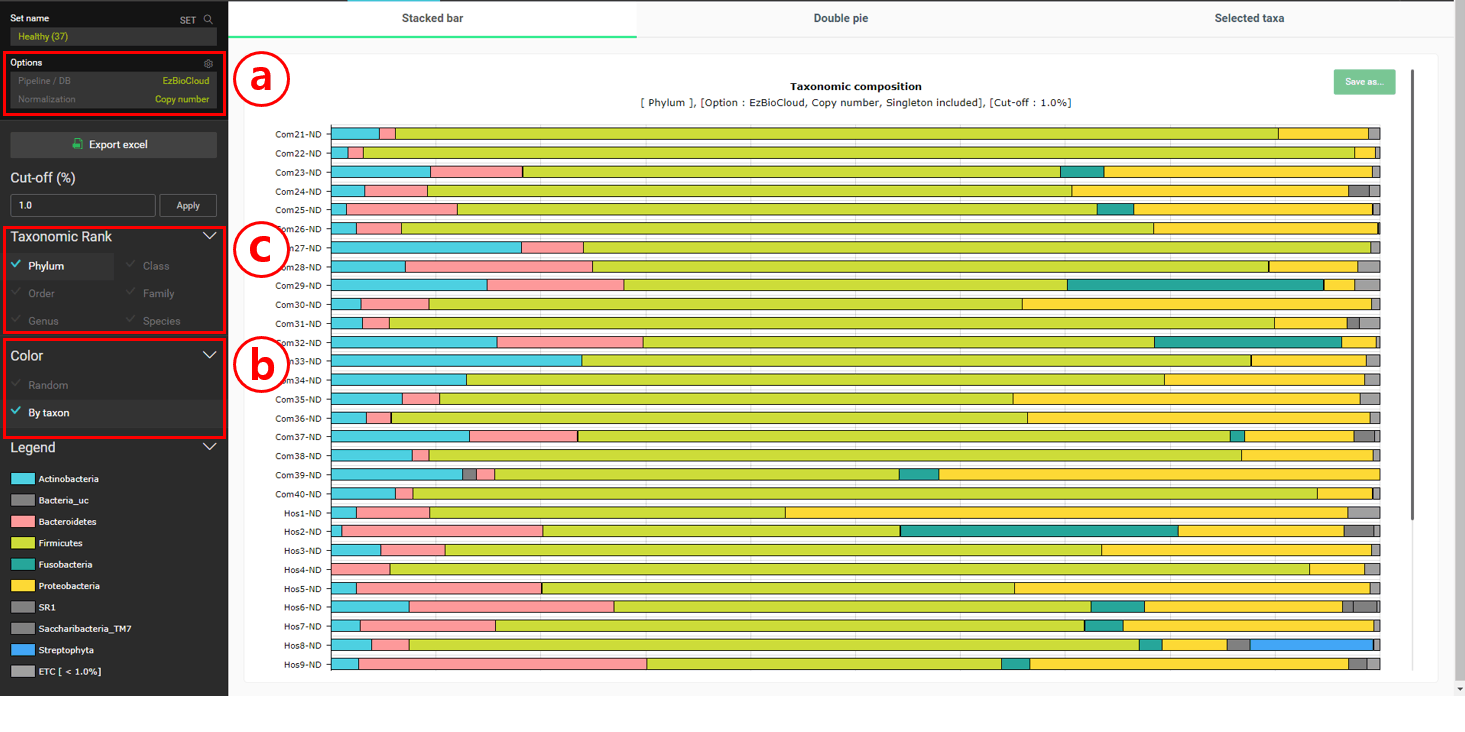
“Composition” 메뉴를 눌러 “Stackced bar” 차트를 선택하세요.
Normalization을 위해 “Copy number”를 설정합니다. 이것은 우리가 taxonomic composition에 대한 통계값을 계산할 때 16S rRNA 유전자의 copy number를 고려할 수 있도록 해 줄 것입니다.
다른 MTP끼리도 같은 taxon은 같은 색으로 표시되도록 “By taxon”에서 색을 설정합니다.
차트에 표시할 taxonomic rank를 변경합니다.
건강한 사람의 경우 phylum level에서 Firmicutes가 가장 풍부한 것으로 보입니다. 종 수준에서는 “Streptococcus pneumoniae group”이 가장 많은 것으로 나타났습니다. “Double pie chart”로 이것을 확인해 봅시다.
이 차트에서는 사용자가 선택한 두 taxonomic rank의 평균적인 taxonomic composition이 주어집니다.
Inner circle을 “Phylum”으로 설정하세요.
Outer circle을 “Species”로 설정하세요.
[Apply] 버튼을 누릅니다.
“Double pie chart”에 선택한 옵션이 반영될 것입니다. 차트에서 taxa의 full name을 보려면 커서를 움직여 주세요.
“Alpha diversity”에서는 set을 구성하는 모든 MTP의 여러 다양성 지수를 볼 수 있습니다. 예를 들면 “Good’s coverage of library” 지수는 모든 MTP에서 100%에 매우 가까우며, 이는 샘플의 시퀀싱 리드 수가 통계적으로 충분하다는 것을 가리킵니다(아래 스크린샷).
“Alpha diversity” 메뉴에서는 ACE, Chao1, Jackknife가 추정된 종의 수를 제시하며, 이는 종 풍부도(species richness)라고 불립니다. “ACE” 탭에서는 각 MTP의 추정된 종의 수(=OTU)가 바 차트로 나열되어 있습니다.
이 숫자는 각 샘플(MTP)의 추정된 종 수를 가리킵니다.
태그를 선택하면 태그가 달린 MTP만 강조될 것입니다. 이 예제에서는 “female”이 태그된 MTP만 강조되어 있습니다. Male이 태그된 MTP에서는 명확한 특징이 발견되지 않았습니다.
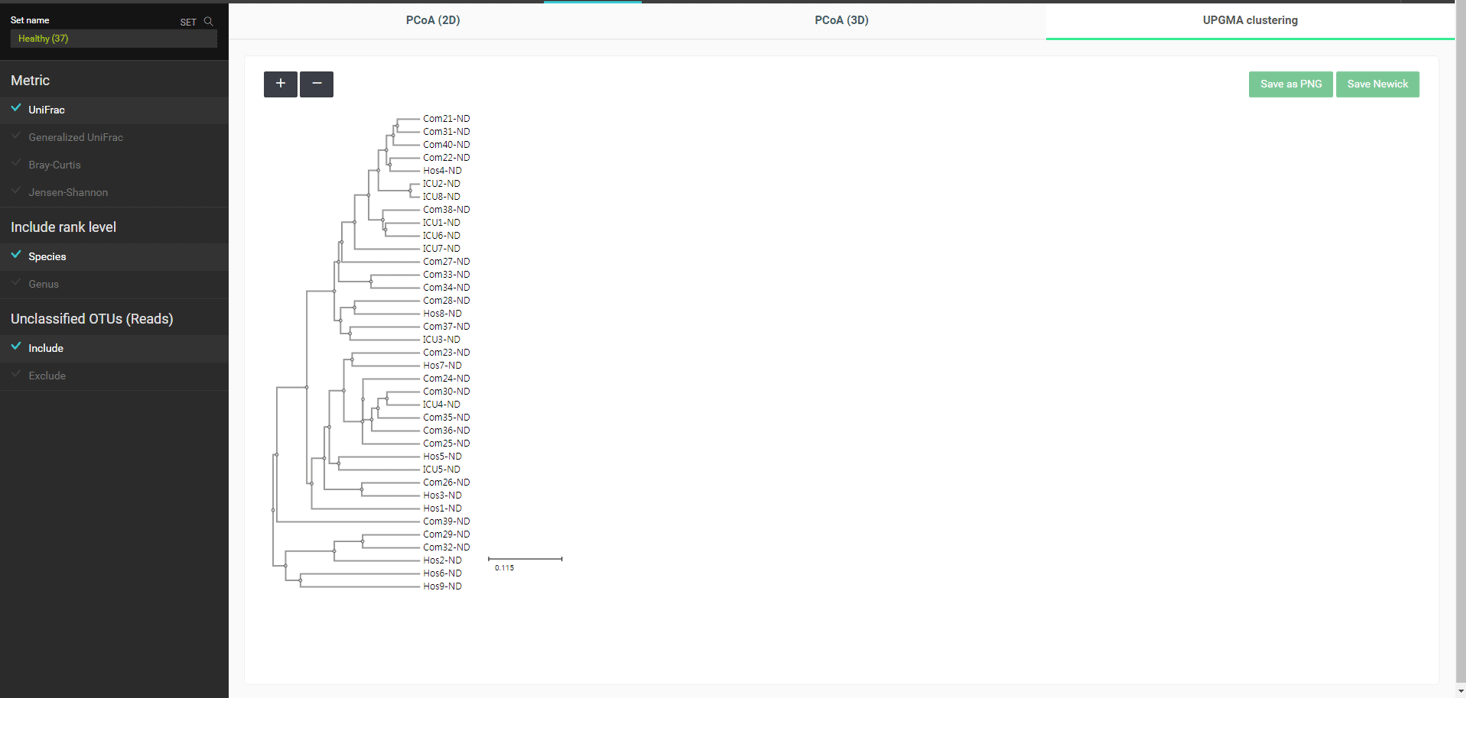
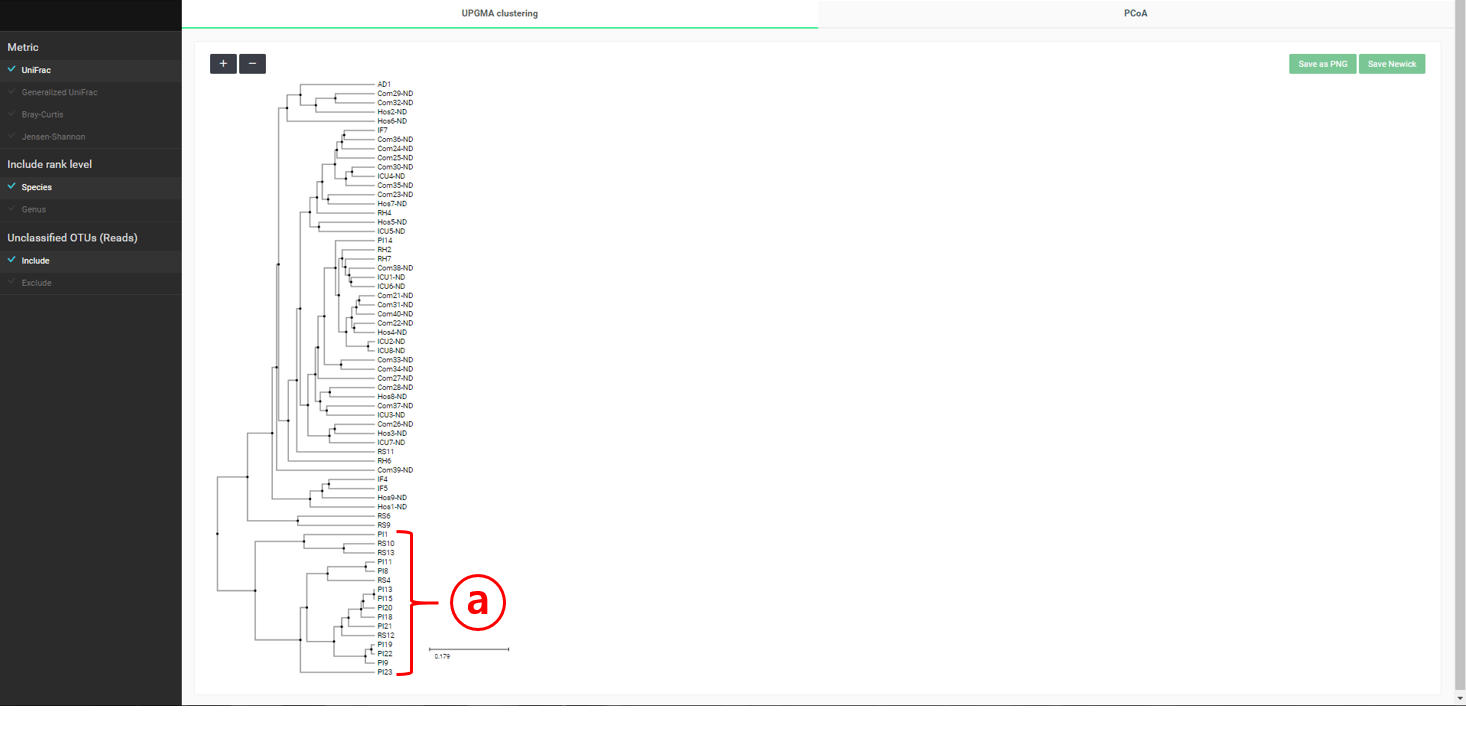
“Beta diversity” 메뉴에서는 다양한 통계 및 시각화 기법으로 샘플 간의 상관관계를 살펴볼 수 있습니다. 가장 널리 사용되는 MTP 간의 거리 측정법은 “UniFrac”입니다. 만들어질 수 있는 모든 쌍(pair)에 대해 거리가 계산되며, 이는 계층 구조 클러스터링이나 principal coordinate analysis (PCoA)에 의한 차원 축소에 활용됩니다. UniFrac distance를 사용하여 “healthy” set의 MTP로 UPGMA clustering한 결과를 아래에서 볼 수 있습니다.
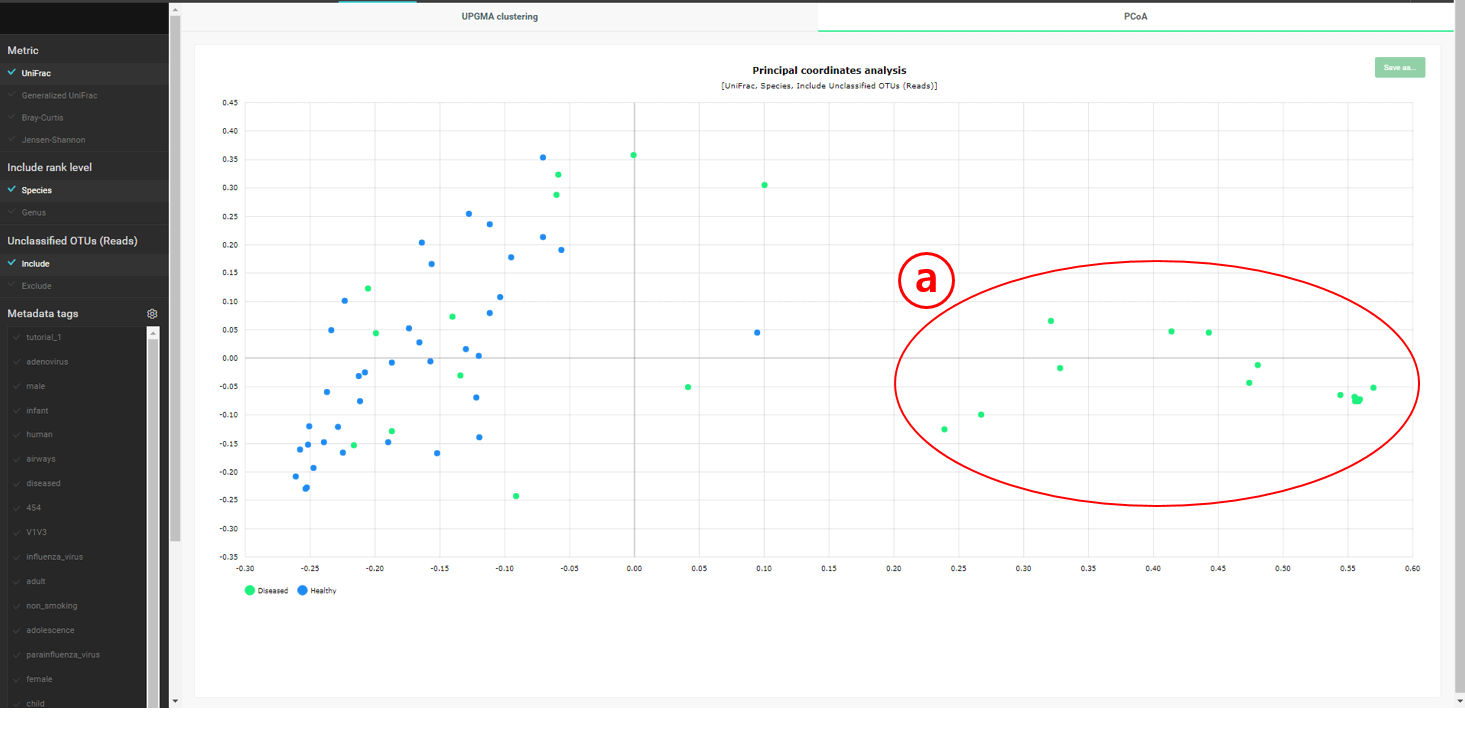
이 데이터 세트는 세 가지 타입의 “healthy” 샘플을 포함하고 있습니다: Com-X-ND (Community subject, non-diseased), Hos-X-ND (Hospital staffs, non-diseased) and ICU-X-ND (intensive care unit staffs, non-diseased). 위의 UPGMA dendrogram에서는 세 그룹이 뚜렷하게 분리되지 않았습니다. 계층 구조 클러스터링이 간혹 편향된 결과를 만들기 때문에 우리는 PCoA에 의한 배열법을 사용해서 이것을 확인합니다. “PCoA (2D)” 탭을 선택해서 건강한 사람 37명의 2차원 분산도(2-dimensional scattergram)를 그릴 수 있습니다. 태그들을 조합하여 선택하면 필요한 MTP를 강조할 수도 있습니다.
1st와 2nd principal component에 기반한 2차원 분산도를 그릴 수 있습니다.
선택한 태그로 MTP간의 상관관계를 탐색할 수 있습니다. 이 차트에서는 community 군이 일관성 있게 모여있지 않으며, 이것은 UPGMA clustering 결과로 확인할 수 있습니다.
여러 MTP set으로 종 다양성 비교하기
비교 연구의 궁극적인 목적은 다양한 특징이 있는 마이크로바이옴을 구분 짓는 바이오마커를 발견하는 것입니다. Tutorial set을 이용한 예제에서는 “환자” 군에만 특별히 존재하는 미생물 종을 찾고자 합니다. 이를 위해 앞서서 두 개의 MTP set을 이미 만들었으며 “healthy”와 “diseased”로 이름을 붙였습니다. 비교 분석 과정은 아래와 같습니다.
“Comparative MTP Analyzer”를 선택합니다.
두 개의 MTP set을 선택합니다.
“Compare taxonomic compositions”를 선택합니다.
[Run]을 클릭하여 비교 모듈을 실행합니다.
“Comparative MTP Analyzer”의 모듈은 “MTP Set Browser”와 매우 유사합니다. “MTP Set Browser”는 세트 안의 개별 MTP에 초점이 맞춰져 있다면, “Comparative MTP Analyzer”는 세트 간의 통계 값을 비교하는 방법입니다.
“Species”를 선택합니다.
“Haemophilus influenzae group”을 선택합니다.
“Haemophilus influenzae group”이 “Diseased” set에만 존재하는 것인지 확인합니다. 환자의 호흡기 샘플에서만 발견되는 종이 적어도 한 종 이상 있다는 것을 발견하였습니다.
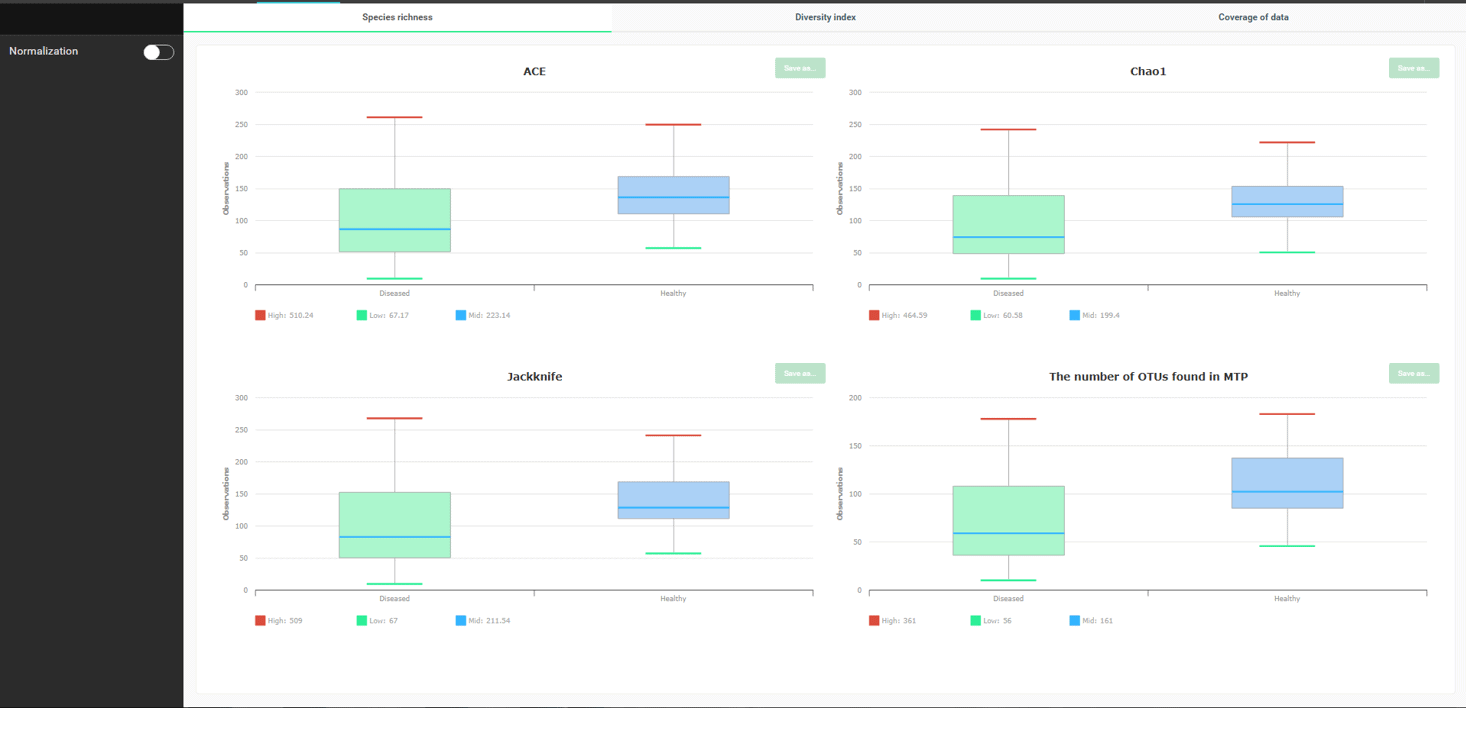
질병에 걸린 사람의 체내 미생물 종의 수와 다양성은 감소한다는 것이 일반적으로 잘 알려져 있습니다. 이것이 사실인지 확인해 봅시다. Alpha-diversity 메뉴를 선택하면 아래와 같은 화면을 볼 수 있습니다.
모든 종 풍부도 지표 (ACE, Chao1, Jacknife)는 “healthy” 군의 미생물 종의 수가 “diseased” 군보다 대체적으로 높다는 사실을 가리키고 있습니다. “The number of OTU found in MTP”는 얼마나 많은 리드가 시퀀싱 되었는 가에 의존하는 결과이므로 너무 많은 의미를 부여하려고 하지 않아도 됩니다.
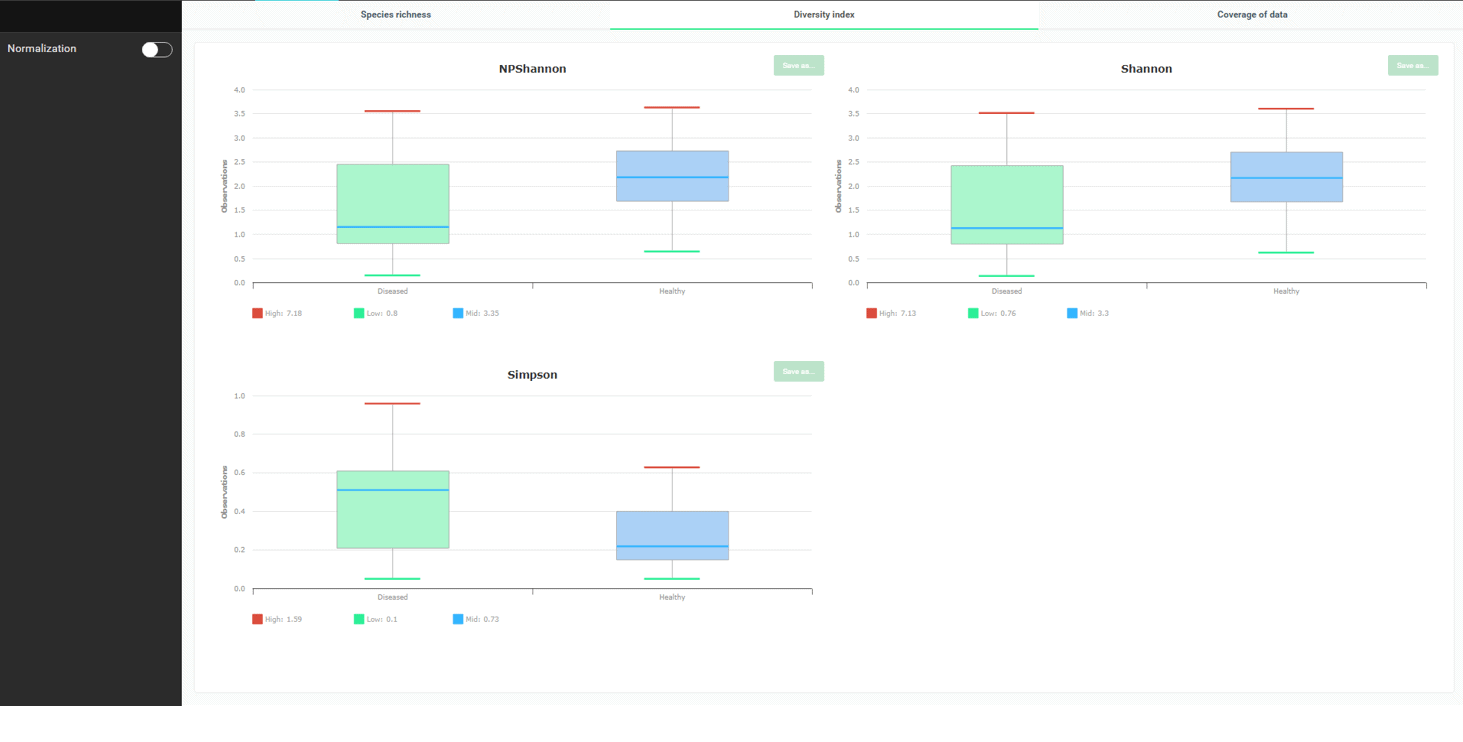
좋습니다. 우리는 이제 healthy 군이 더 많은 미생물 종을 가지고 있다는 것을 알았습니다. 종 다양성과 종 균등도는 어떨까요? 확인하기 위해 “Diversity Index” 탭으로 가 봅시다.
세 개의 다양성 지수가 있으며, 이들은 healthy 군이 더 많은 종 다양성을 나타낸다는 것을 명확하게 보여주고 있습니다. NPShannon과 Shannon의 값이 클수록 다양성이 높아진다는 것에 유의하여야 합니다. Simpson 지수는 반대로 값이 클수록 다양성이 낮아집니다.
상관관계를 밝히는 다중 MTP set 비교
“Comparative MTP Analyzer”의 “Beta-diversity” 메뉴를 시작하면 두 개의 탭이 나타납니다. “UPGMA clustering”은 “healthy” set과 “diseased” set 양 쪽 모두의 MTP를 포함하는 dendrogram을 그려 줍니다(아래 참조).
“Diseased” 군 만을 포함하는 클러스터가 밝혀졌습니다. 이 클러스터는 아래의 “PCoA” plot에도 명확하게 나타납니다.
유의미한 taxa (바이오마커) 찾기
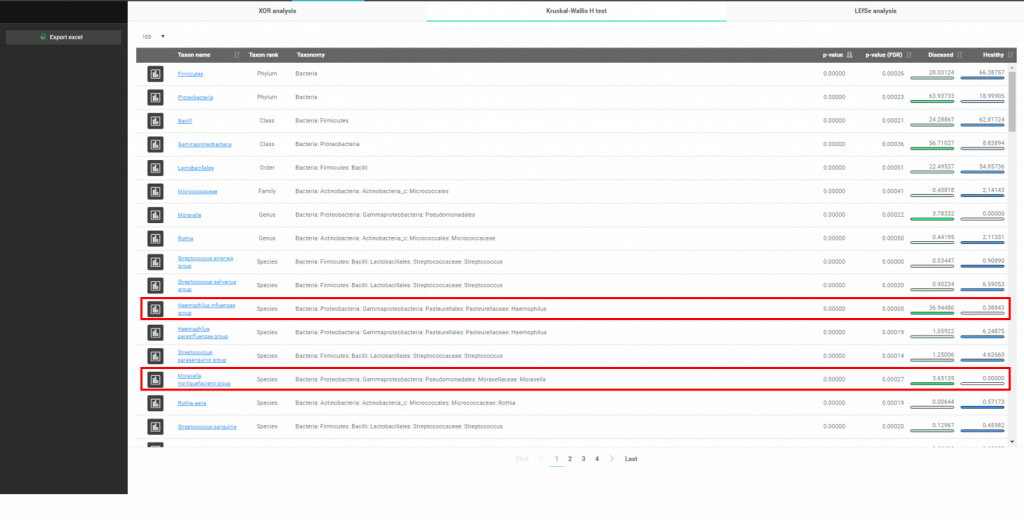
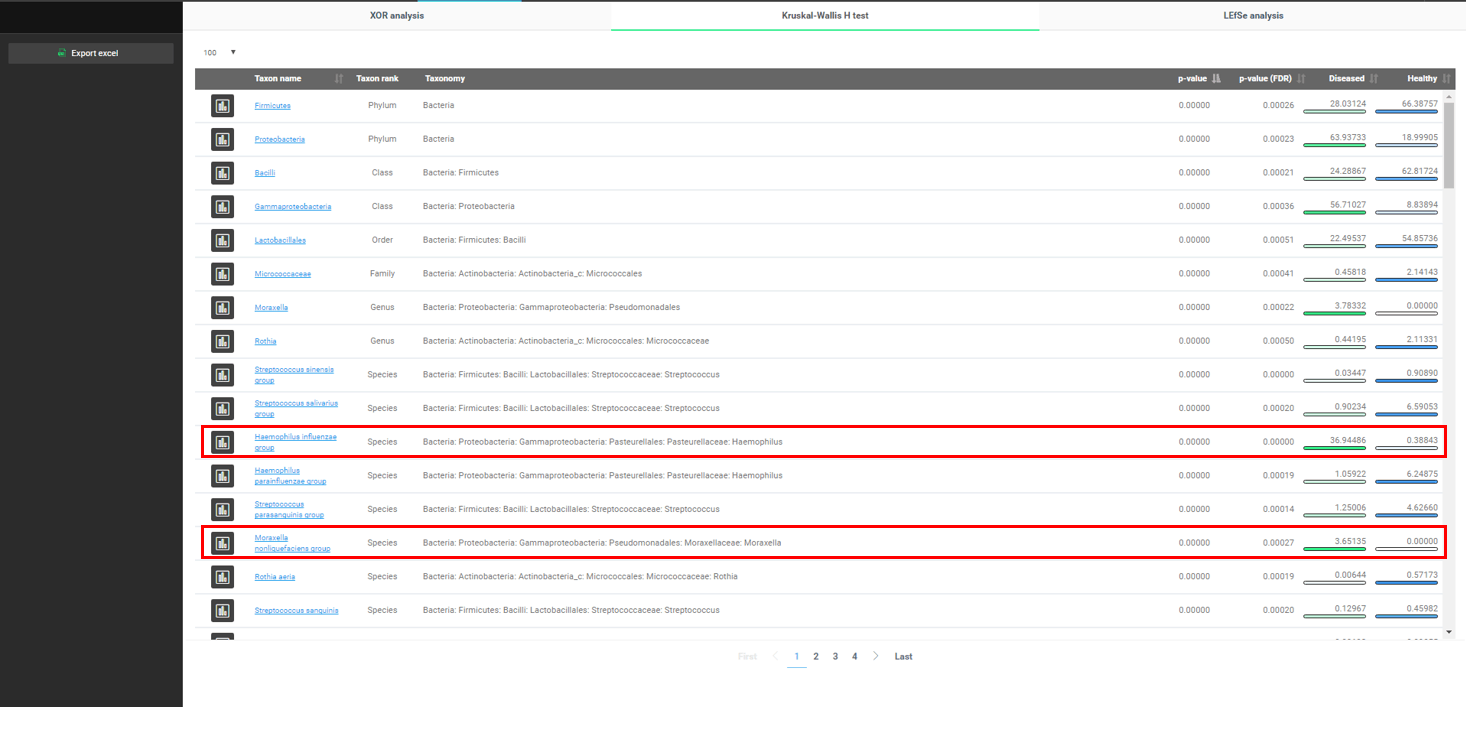
마지막으로, 우리는 두 조건 간에 어떤 주요한 차이가 있는지를 데이터로 조사해 볼 것입니다. EzBioCloud는 바이오마커를 발견하는 여러 가지 방법을 제공합니다. 여기서는 “Kruskal-Wallis H test”를 사용하여 호흡기 질환과 어떤 미생물이 연관되어 있는지를 찾아보겠습니다. “Biomarker Discovery” 메뉴를 선택합시다.
Phylum부터 species까지 유의미한 taxa가 p-value 값에 따라 정렬되어 있습니다. 앞에서 우리가 이미 발견했던 “Haemophilus influenzae group”이 목록에 나타난 것에 주목합시다.
흥미롭게도 “Moraxella nonliquefaciens group”도 “diseased” 군에서만 발견되고 “healthy” 군에서는 발견되지 않았습니다.
마치며
연구자들이 적절한 생물정보학 도구와 전산 환경을 갖춘다면 마이크로바이옴 데이터를 다루는 방법은 무한할 정도로 많습니다. EzBioCloud의 클라우드 환경은 비교 분석, 시각화, 데이터 마이닝에 필요한 즉각적인 분석 시스템을 제공하기 위해 노력하였습니다. 이 tutorial을 즐겁게 따라오셨기를, 그리고 EzBioCloud의 특별한 사용자 인터페이스와 친숙해 지셨기를 바랍니다.
알림
이 tutorial은 Dr. Jon Jongsik Chun (Seoul National Univ/ChunLab, Inc)과 Suyeon Hong (Yale Univ)에 의해 작성되었습니다.
이 Tutorial은 EzBioCloud(http://www.ezbiocloud.net/)의 비교 유전체(comparative genomics) 분석 이용 방법을 설명하기 위해 작성되었습니다. 여러분의 이해를 돕기 위하여 잘 알려진 병원성 미생물인 Vibrio cholerae (콜레라 원인균) 를