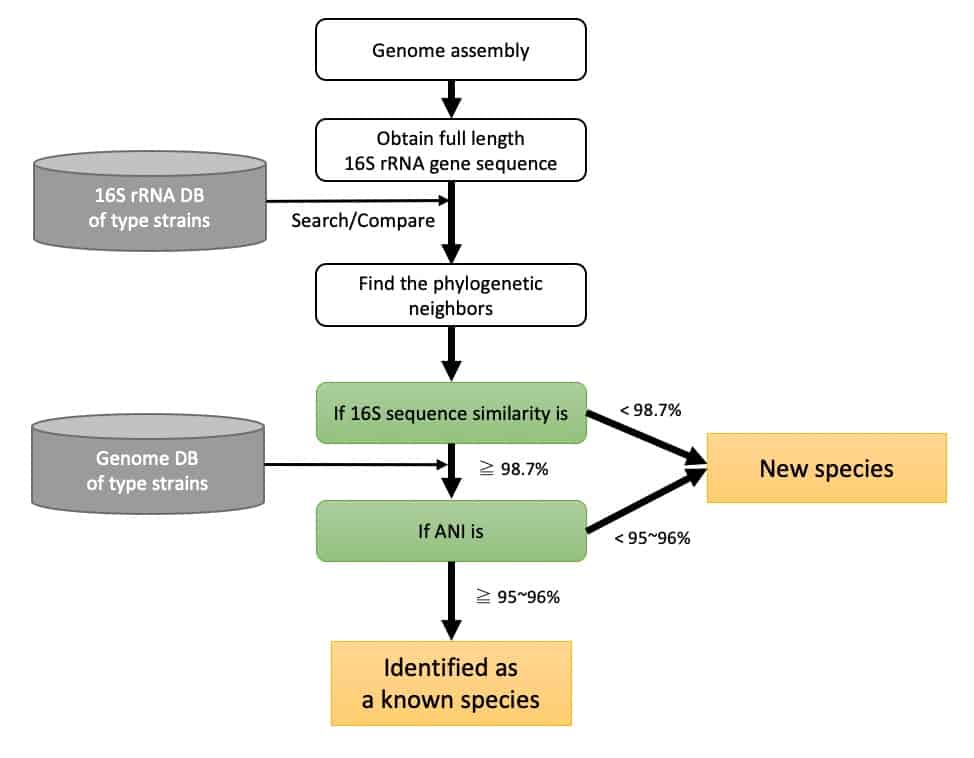
Species Concept-based Identification
The bacterial species concept is now based on a direct comparison of genome sequences, so a species concept-based identification scheme can similarly be built using genome sequence data. This process involves two steps: (1) selection of phylogenetically close species using a fast search engine and (2) calculation of average nucleotide identity (ANI) to the chosen species. The generally accepted ANI cutoff for species boundary is 95~96 %. These general standards and procedures for taxonomic purposes have been proposed and should give a good overview of the species concept-based identification scheme. [Chun et al., 2018].
Genome-based bacterial taxonomy
The EzBioCloud team / Last edited on Mar. 25, 2018
TrueBac TM ID algorithm
Algorithm for identification of a bacterium using its 16S rRNA gene sequence
The most critical measurement for 16S-based species identification is pairwise sequence similarity. However, different sequence alignment algorithms may produce different similarity values. Therefore, it is important to use a taxonomically valid algorithm for alignment and similarity calculation. It is ideal if we calculate all similarities between the isolate and all type strains of the known species. This is doable, but not efficient as it will take very long for computing all pairs (>70,000) while we only need the values that are close enough (i.e., species with >97% similarity). For this reason, a two-step approach is devised for the TrueBac ID-16S service. It is the same as the one used on our public [Identify] service (www.ezbiocloud.net), except that the reference database used in TrueBac ID-16S is more stringently curated. [Learn more].
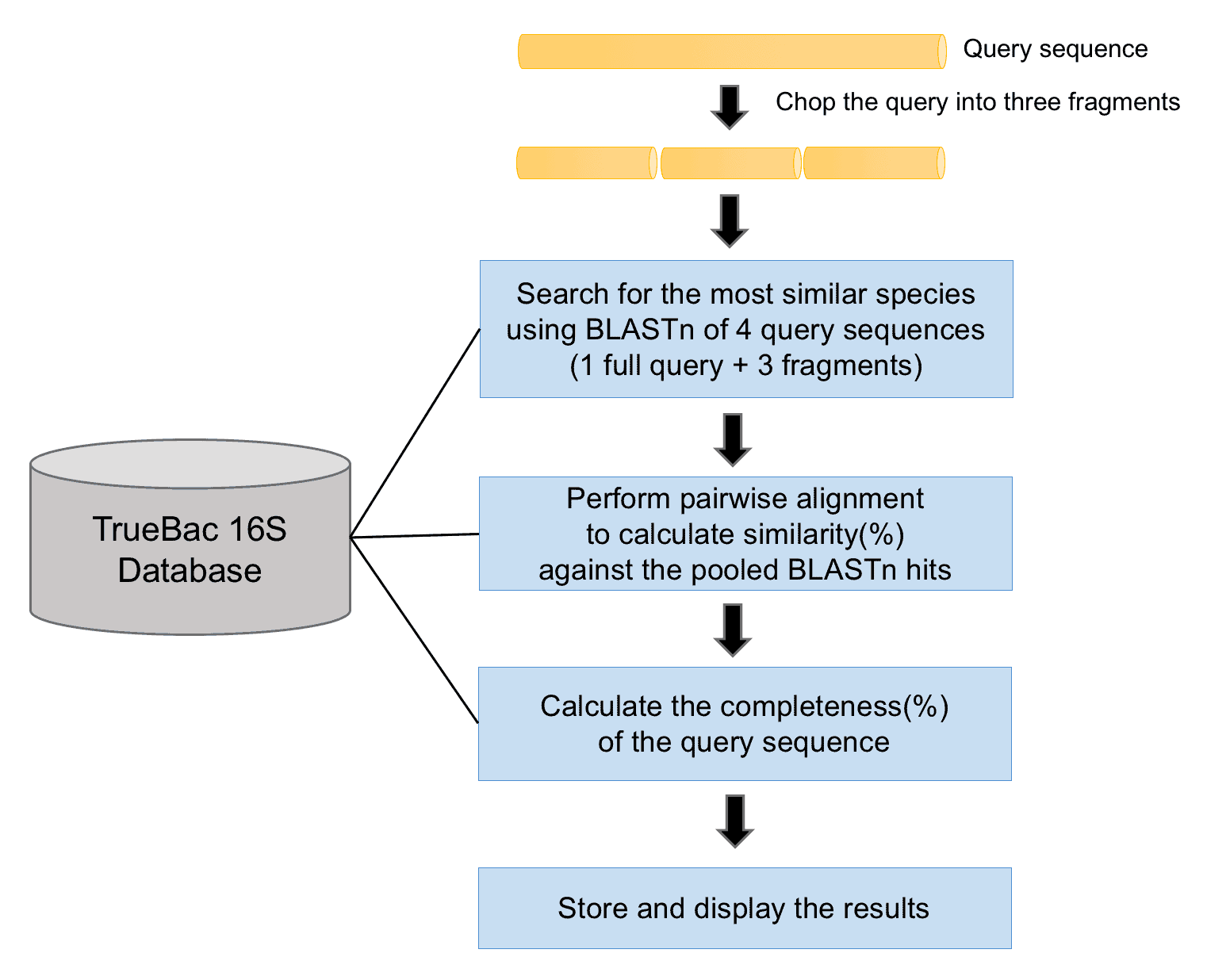
TrueBac ID-16S algorithm (the above figure) performs the following steps:
- The query sequence is chopped into three fragments of equal length. If the length of the query sequence is < 1000 bp, the query is chopped into two fragments. If the length of the query sequence is < 500 bp, the query will not be chopped.
- The original full-length query and the fragmented sequences, four sequences in total, are used as the query sequence for a BLASTn-based search against the TrueBac 16S Database. Using the different parts of the query sequences in the BLASTn search ensure the correct identification of all potentially similar reference sequences. Fifty hits are collected from each of the four BLASTn searches and combined. Because there are always duplicated hits, the final hit list contains much less than 200 hits.
- A robust pairwise sequence alignment (Myers and Miller, 1988) is carried out between all pairs, that is, the query sequence against all BLASTn hit species identified in the previous step. The alignment algorithm used in TrueBac ID-16S service is same as the one used in defining the 16S cutoff (98.7%) for species definition (Kim et al., 2014) and used in the highly cited EzBioCloud (formerly EzTaxon) service. For more details about 16S similarity calculation, please read this article. Please note that BLASTn identity values are not used for taxonomic purposes [Learn more].
- The completeness(%) of the query sequence is calculated [Learn more]. For example, 50% completeness means that the query sequence covers only half of the full-length 16S gene. The taxonomically meaningful 16S sequence similarity was proposed on the basis of full-length sequences. Therefore, similarity values based on partial sequences should be interpreted carefully.
- Finally, the hit species are sorted by the 16S similarities and displayed as a table and stored. The completeness (%) of the query sequence is also provided.
Interpretation of 16S similarity values should be carefully done. For example, Bacillus cereus shows >99.8% 16S similarity to about ten species [Learn more], implying that very similar 16S sequence does not always mean that the isolate belongs to the hit species.
Algorithm for identification of a bacterium using it’s whole genome sequence
A bacterial isolate can be confidently identified at the species level using genome sequence information (Richter & Rosselló-Móra, 2009; Chun & Rainey, 2014; Chun et al., 2018).
If strain 1 belongs to species A, the necessary conditions are:
- The genome sequence of the type strain of species A must be available.
- The average nucleotide sequence identity (ANI) value between the genomes of the strain 1 and type strain of species A should be higher than the proposed cutoff for bacterial species definition, i.e., 95~96%.
Ideally, ANI values are calculated for the genome sequence of the isolate against the genome of type strains of all known species. This would require an enormous computing resource and is not efficient as we are only interested in the species identification. In other words, only closely related species would matter. For this reason, TrueBac ID-Genome adopted a two-step approach where, first, the potential hit species are identified using four-way searches which are then used to compute ANI values with the query genome. Sometimes, a good-quality sequence cannot be extracted from the final genome assembly. Therefore, in addition to the 16S gene, the recA and rplC genes are used for searching the potential neighboring species. The RplC is a ribosomal protein whereas RecA is not. They are members of the recently revised bacterial core gene set [Learn more].
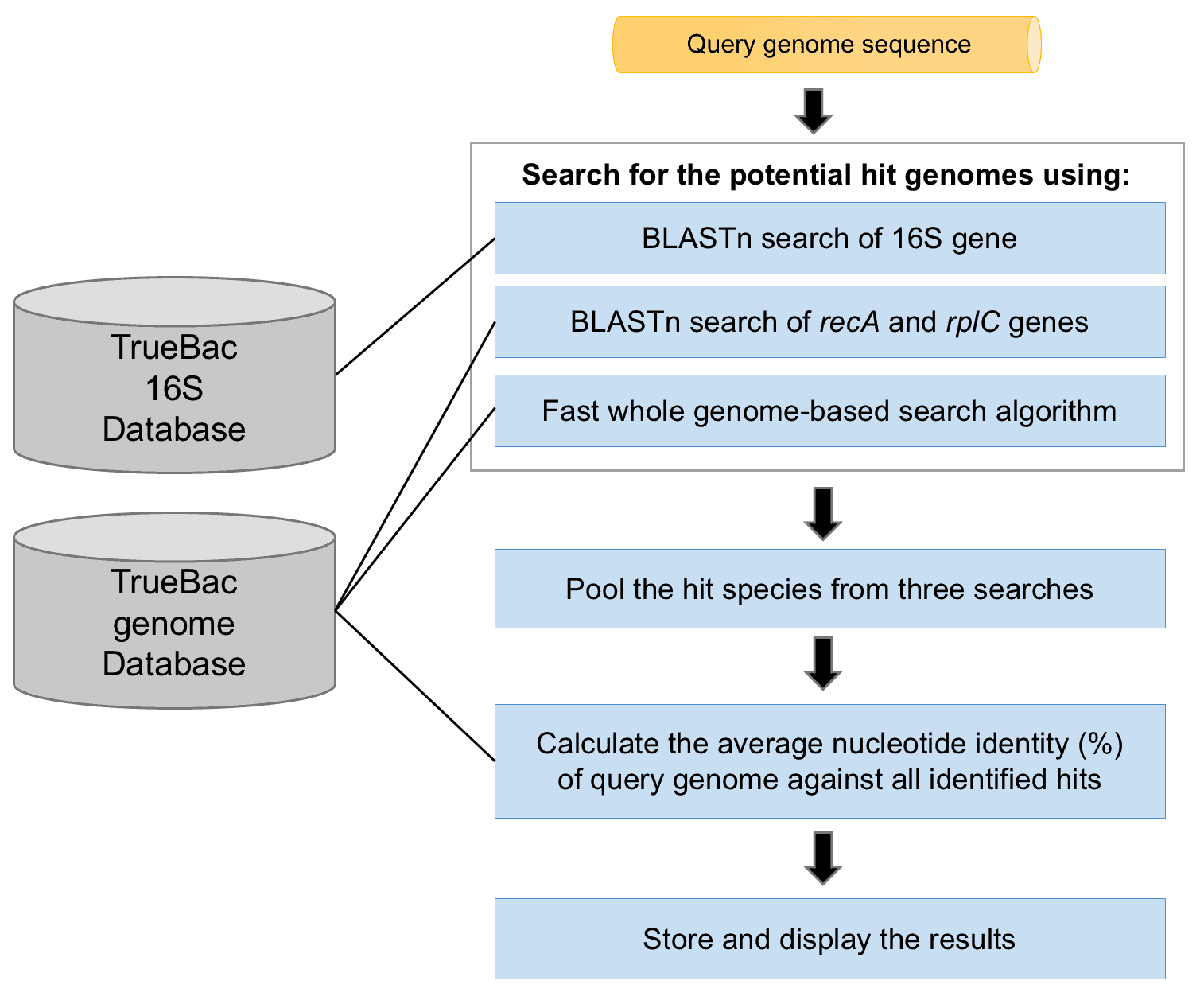
The TrueBac ID-Genome algorithm (figure above) performs the following steps:
- The query genome is used to find the potential hit species against the TrueBac DB using the following search engines:
1.1. If the query genome contains 16S sequence, it will be used to search against the TrueBac DB using BLASTn program.
1.2. If the query genome contains recA gene sequence, it will be used to search against the TrueBac DB using BLASTn program.
1.3. If the query genome contains rplC gene sequence, it will be used to search against the TrueBac DB using BLASTn program.
1.4. Additionally, at least one search algorithm that utilizes whole-genome information is used. At present, the MASH tool (Ondov et al., 2016) is included in our pipeline. - The potential hit species identified in the step 1 are pooled and used for computing average nucleotide identity using the MUMmer tool (ANIm; Richter & Rosselló-Móra, 2009)
- The decision for species-level identification is made by considering ANIm and 16S similarity values.
The TrueBac ID-Genome algorithm (figure above) decision making process:
- If there is a known species (species with valid name or genomospecies) with ≥95 % ANI, the decision is made as “Identified correctly to that species.”
- If there are no known species with ≥95 % ANI, the decision is made as “sp. nov.” (meaning novel species).
- If there is an ambiguity in making an identification call, the decision is made as “sp.” (e.g., Bacillus sp.). It includes the following cases.
3.1. The genome sequence(s) are not available for the type strains of some of the potential hit species. In this case, the result of identification can be updated later when TrueBac DB is updated with that missing genomes in future.
3.2. The query genome shows identical or very similar ANI values to two or more species. In most cases, the latter species belong to the same species. In other words, they are synonyms but the necessary taxonomic change (i.e., combining them into a single species) has not proposed yet.
Please note that the TrueBac DB contains >2,000 genomospecies [Learn more]. If your query genome is identified as one of these, it means that you have a novel species. Genomospecies are included in the database and are being expanded constantly to provide users the way of tracking isolates belonging to novel species. For example, CP015110 (a genome sequence deposited in NCBI) represents a novel species in the genus Acinetobacter so it was named the genomospecies CP015110_s in TrueBac DB. If a user isolates multiple strains belonging to this species, they will be identified as “CP015110_s” instead of “Acinetobacter sp. nov.” by TrueBac ID-Genome service. In this way, most, if not all, isolates can be properly classified and organized easily according to the species.
The EzBioCloud team / Last edited on May 01, 2018
TrueBac ID Demonstrations
TrueBac ID has been designed to definitively identify bacteria using whole genome sequence data. Here we have run TrueBac ID on some publicly available data to highlight the accuracy of the system. Because taxonomy and the TrueBac Reference Database are constantly being updated, the data presented here are those at the time of analysis.
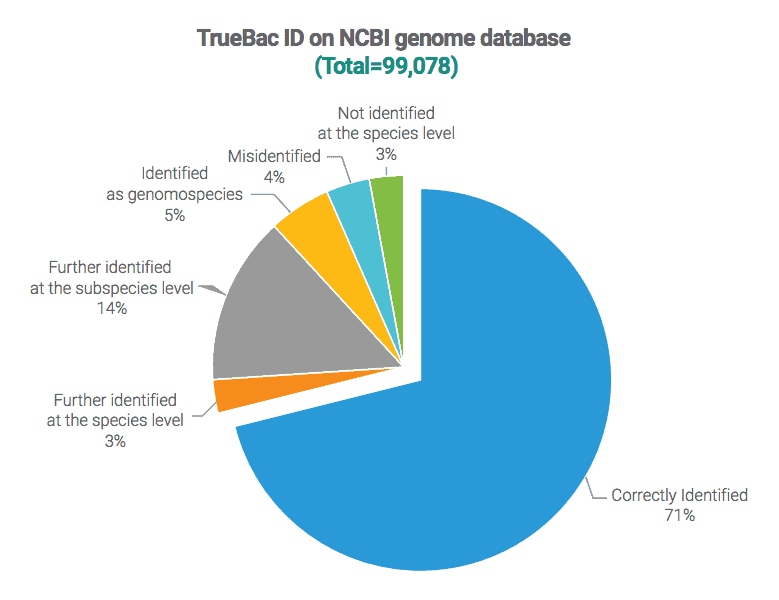
Case #1: NCBI bacterial genome database
Input dataset contains 99,078 bacterial genomes from pure cultures (excluding metagenome and single cell assemblies). Contaminated genomes were also excluded by the ContEst16S tool. All identification results of TrueBac ID are provided at www.ezbiocloud.net. The identification was carried out on May 15, 2018.


Case #2: An unbiased collection of clinical isolates
A team at the University of Washington Medical Center published the genome data of >1,200 bacterial strains isolated from an Intensive Care Unit for a year (Roach et al., 2015; PLOS Genetics 11:e1005413). TrueBac ID was used to re-analyze the same dataset. The identification was carried out on May 15, 2018.


Identification of clinical isolates from ICU for a year using TrueBac ID-Genome

Detailed identification results are available here.
Case #3: Accurate identification of a gut bacterium fails using MALDI-TOF and other conventional methods
A team at Harvard University isolated a potential therapeutics strain from human gut. This strain could not be identified by MALDI-TOF or other conventional methods, so it was tentatively proposed as a novel species ‘Clostridium immunis‘ (Nature 2017; 14;552(7684):244-247) . TrueBac ID successfully identified this strain as Clostridium symbiosum by 98.48% Average Nucleotide Identity (ANI).

Categories of TrueBac ID results against the original species/subspecies designations in the database or publications
Further identified as the species level:
The original name of the genome has the correct genus name but does not contain specific epithet. In this example, Sulfitobacter sp. NAS-14.1 (GCA_000152645.1) is identified as Sulfitobacter pontiacus.
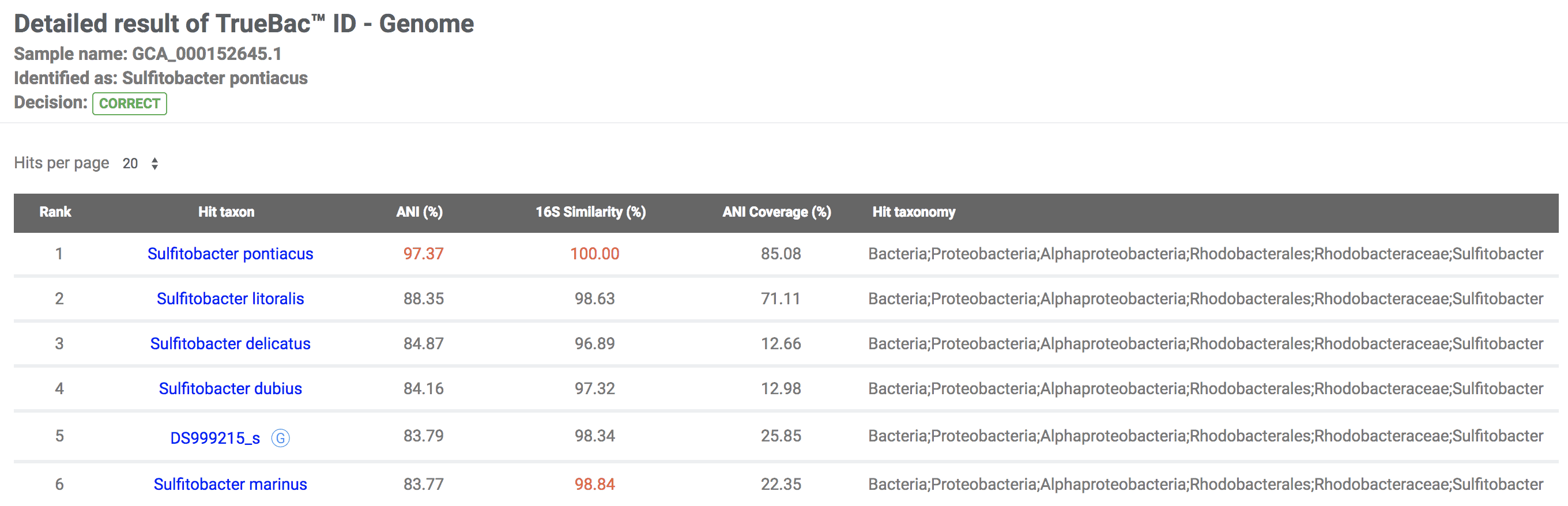
Further identified as the subspecies level:
The original name of the genome has the correct species name but does not contain subspecies name. In this example, Pasteurella multocida FDAARGOS_384 (GCF_002393385.1) is further identified to Pasteurella multocida subsp. septica.

Identified as a genomospecies:
Genomospecies is a potentially novel species and tentatively named in EzBioCloud/TrueBac databases [Learn more]. Actinomyces odontolyticus ATCC 17982 (GCF_000154225.1) is not a strain of Actinomyces odontolyticus but represents a novel species which we named a genomospecies (DS264586_s).

Misidentified:
Bacillus cereus ATCC 10987 (GCA_000008005.1) is identified as Bacillus pacificus with 99.84% ANI. It is clearly not a strain of Bacillus cereus as ANI value to B. cereus type strain is only 91.96%.
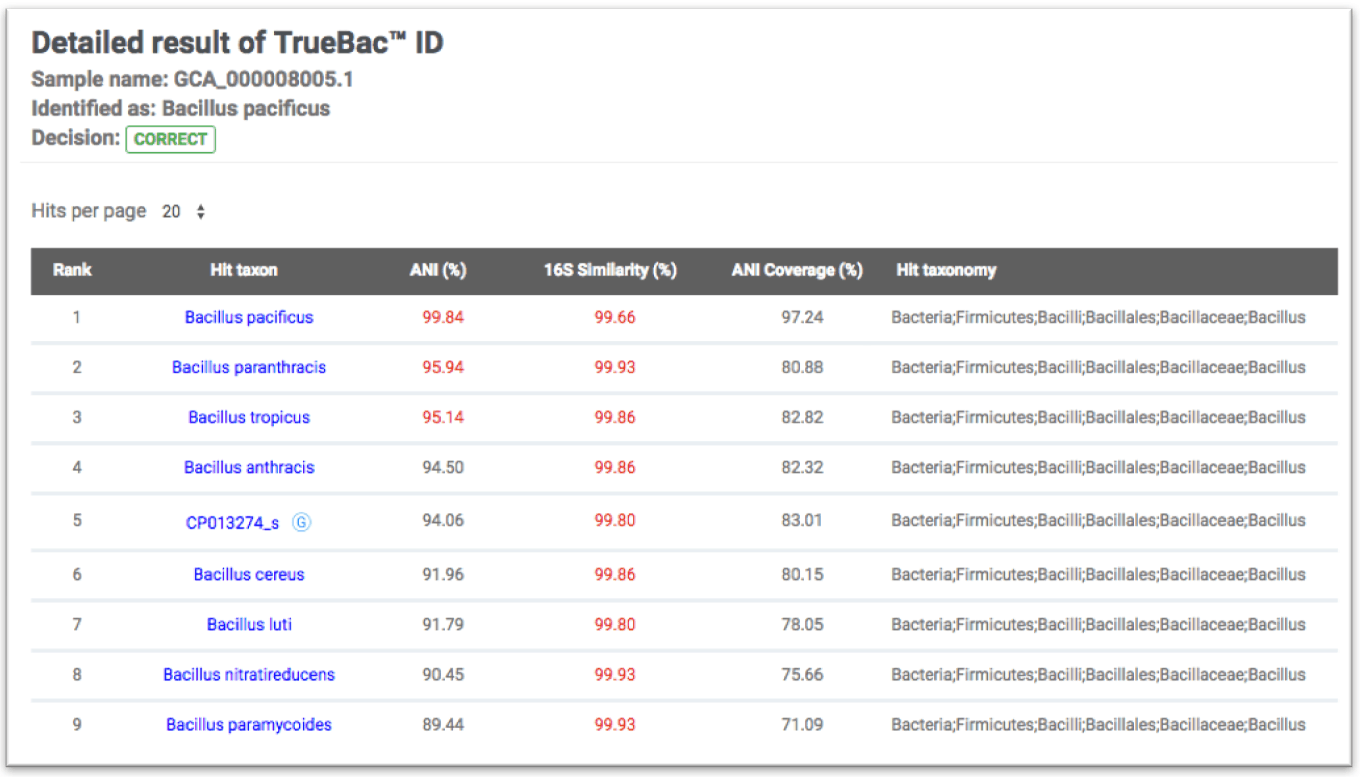
Not identified at the species level:
Because either it is a novel species or there is no sufficient reference genome data, the genome cannot be identified to a known species with confidence. Haemophilus parainfluenzae strain 1209_HPAR (GCA_001053035.1) is identified as a novel species. In this example, the closest known species is Haemophilus parainfluenzae.
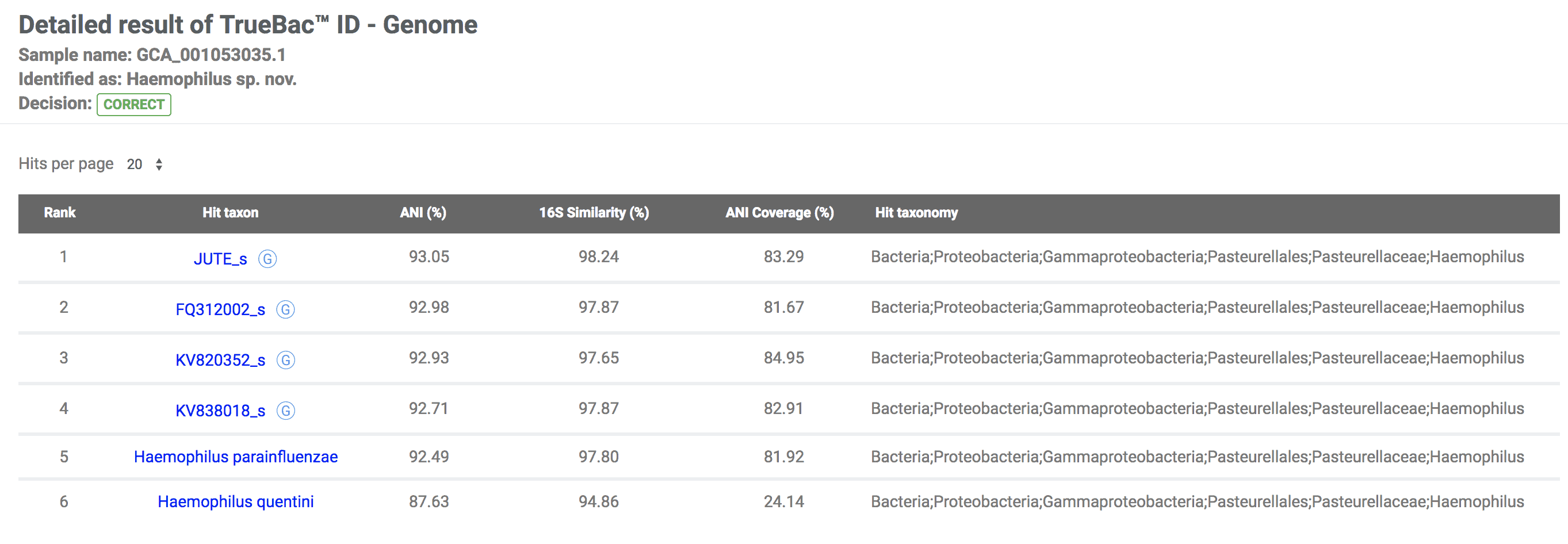
The EzBioCloud team / Last edited on May 22, 2018

