What is the UBCG?
UBCG stands for the up-to-date bacterial core gene. It is a method and software tool for inferring phylogenetic relationships using a bacterial core gene set that is defined by up-to-date bacterial genome database.
This document is for version 3. If you have an older version, please download and install the latest version.
How to cite the UBCG pipeline
If you use this tools, please cite the following:
Na, S. I., Kim, Y. O., Yoon, S. H., Ha, S. M., Baek, I. & Chun, J. (2018). UBCG: Up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J Microbiol 56, (in press). [View only version]
Gene set used in the UBCG pipeline
The most widely employed method for genome-based phylogenetic tree reconstruction is using the core gene set. The core gene set can be defined as
- Genes that are present in the majority of species, if not all
- Genes that are present in a single copy (likely orthologous but not paralogous)
The number of core genes varies depending on the scope of a target taxon. If you generate a phylogenetic tree for a species, the core gene set may consist of up to thousands of genes. However, to cover any taxa in the domain Bacteria, the core gene set should be restricted to the highly conserved ones (Bacterial Core Gene [BCG]).
Because the number and taxonomic coverage of complete genome sequences in the public database are not perfect, the number of BCG set varies over time.
Here, we compiled the latest bacterial core gene set, named UBCG, using the largest dataset ever (1,429 complete genome sequences, a single genome per a species, covering 28 phyla). The current UBCG set consists of 92 genes whose details are given here.
Concept of the UBCG pipeline
We designed the pipeline for users to handle hundreds of genomes, if not thousands. Here, the concept behind our design is briefed to help you understand and maximize the utility of our pipeline.
- All UBCG sequences extracted from each genome sequence are stored in a single file (*.bcg). This file also contains a label with full information about the strain (e.g. Escherichia coli K12 MG1665) and other details (e.g. database accession). Once a bcg file is generated, it can be used for different analyses. This allows users to change the labels in the phylogenetic trees.
- A run is carried out using a set of bcg files of user’s choice. For this, selected bcg files are saved in a single directory, then the UBCG pipeline will align each of the core genes, concatenate them, filter aligned positions, and calculate phylogenetic trees and gene support indices (GSIs).
- If a user wants to run the pipeline for another set of bcg files, store the desired bcg files in bcg directory and re-run the pipeline. In other words, the set of bcg files to be analyzed together is controlled by the content of a directory holding bcg files.
Installation
- The lasted version is available at https://www.ezbiocloud.net/tools/ubcg.
- It has been tested on Linux and Mac OS X 10 or higher. MS Windows is not supported due to the external programs used. Please use a virtual Linux machine such as the Virtualbox.
- Unzip the UBCG.zip file in the desired directory.
File formats used in UBCG tool
| File | Function |
| extension | |
| *.bcg | The files with *.bcg extension are of JSON format and contain all extracted UBCG gene sequences with metadata (data about data). This file is a text format and readable by any text editor. So, you can extract sequence information and edit metadata, if necessary. bcg is designed to hold all necessary information about genome and strain. |
| *.fasta | FASTA is a standard file format for holding genome sequences. In UBCG tool, all fasta files containing genome sequences should be converted to bcg files before generating multiple alignments and inferring phylogenetic trees. The results of multiple alignments are also written as fasta format files. |
| *.nwk | Newick is a standard format for phylogenetic trees. |
| *.trm | A JSON format file containing Newick-format trees and metadata of individual core gene trees and a UBCG tree. |
| *.log | A log file is a text format file that contains detailed information about the pipeline run. |
A typical structure of directories
- The program’s root directory should contain the “UBCG.jar” file and the “programPath” file [Learn more] that contains the location information of the external software tools.
- “fasta” directory contains the FASTA format files holding example genome/contig sequences.
- “bcg” directory contains JSON format files (=*.bcg) holding UBCG gene sequences with metadata.
- “output” directory contains all output files generated by the UBCG tool. Within the “output” directory, results of each run are stored in the separate directory (defined by “-prefix”).
Installing external programs
- The following programs should be installed in advance. The locations of programs should be written in “programPath” file.
- PRODIGAL
- For gene-finding
- https://github.com/hyattpd/prodigal/releases/
- HMMER3
- For identifying UBCG genes
- http://hmmer.org/
- We only need “hmmsearch” program.
- FastTree
- For drawing maximum likelihood tree
- http://www.microbesonline.org/fasttree/
- RAxML
- For drawing maximum likelihood tree
- https://sco.h-its.org/exelixis/web/software/raxml/index.html
- You may also install and use other tools for phylogenetic inferences. Since we provide multiple-alignment files, any phylogenetic inference program can be used to generate phylogenetic trees from UBCGs.
Running UBCG pipeline
Step 1: Converting genome assemblies or contigs (fasta) to bcg files
- Command: java -jar UBCG.jar extract
- This command converts a fasta file to bcg file using prodigal and hmmsearch tools.
- You are required to designate the following parameters:
- -i : path of an input FASTA file containing genome assembles.
- -bcg_dir : directory for all bcg files. The name of bcg file will be same as the fasta file. (e.g. -bcg_dir bcg)
- -label : full label of the strain/genome. It should be encompassed by single quotes (e.g. -label “Escherichia coli O157 876”).
- The followings are optional, but useful metadata
- -taxon : name of species (e.g. -taxon “Escherichia coli”)
- -strain : name of the strain (e.g. -strain “JC 126”)
- -type : add this if a strain is the type strain of species or subspecies (e.g. -type)
- -acc : accession of a genome sequence. Usually, NCBI’s assembly accession is used for public domain data.
- -uid : this is a unique integer id. If you do not designate, one will be automatically generated for you. Ignore this when you are not sure about this field.
- The content of bcg files (for example, gene sequences) can be viewed (as CSV format that is readable by Microsoft Excel or Google spreadsheet) by using the following command:
- java -jar UBCG.jar view -i <a bcg file name>
- java -jar UBCG.jar view -d <directory containing bcg files>
Step 2: Generating multiple alignments from bcg files
- Place all bcg files that you want to include in the analysis into a single directory by copying desired bcg files.
- Command: java -jar UBCG.jar align
- You are required to designate the following parameters:
- -bcg_dir directory for bcg files that you want to include in the alignment.
- Optional parameters:
- -out_dir directory where all output files will be
- -a <string>: alignment method (default : codon).
- nt : nucleotide sequence alignment
- aa : amino acid sequence alignment
- codon : codon-based alignment (output is nucleotide sequences, but alignment is carried out using amino acid sequences).
- codon12 : same as “codon” option but only 1st and 2nd nucleotides of a codon are selected. The 3rd position is usually of high variability.
- -t <integer> : number of threads to be used (default : 1)
- -f <integer> : set a filtering cutoff for gap-containing positions (default: 50)
- Enter 0~100
- 0 to select all alignment positions
- 100 to select positions that are present in all genomes
- 50 to select positions that are present in a half of genomes
- -prefix <string>: a prefix is to appended to all output files to recognize each different run. If you don’t designate, one will be generated automatically.
- e.g. john_115, mycoplasma_1
- -gsi_threshold: Threshold for Gene Support Index (GSI). 95 means 95%. (default = 95)
- -raxml : Use RAxML for phylogeny reconstruction (Default: FastTree). Be aware that RAxML is much slower than FastTree.
- -zZ : Make zZ-formatted files. This additionally creates fasta/nwk files with zZ+uid+zZ format for the names of each genome
- Examples of typical runs
- java -jar UBCG.jar align -bcg_dir bcg -prefix mytest1 (align and draw trees with bcg files in “bcg” directory and save all results in “output/mytest1” directory.
- Output files will be generated in output directory (for default) or the directory that you designated with the following name: (assuming that the prefix is mytest1)
- nwk files can be viewed by MEGA, FigTree and other tree viewers. MEGA was tested for displaying Gene Support Index (GSI) on the branches of phylogenetic trees.
- mytest1.log = a text file containing logs (what happened during execution of program)
- mytest1.UBCG_concat.codon.label.nwk = A Newick file based on UBCG gene set, codon alignment, 50% filtered, labeled with full label
- mytest1.UBCG_gsi(92).codon.50.label.nwk= A newick file based on UBCG + Gene Support Index (GGI) values with full label
- mytest1.concat.codon.50.label.fasta = A FASTA file containing multiple alignments of UBCG genes, codon aligned, 50% filtered with full label
- mytest1.concat.codon.50.zZ.fasta = A FASTA file containing multiple alignments of UBCG genes, codon aligned, 50% filtered with zZ+Unique id+zZ
- mytest1.secY.codon.50.label.nwk = A newick file based on a single gene (secY), codon aligned, 50% filtered with full label
- mytest1.secY.codon.50.zZ.nwk = A newick file based on a single gene (secY), codon aligned, 50% filtered with zZ+Unique id+zZ
- mytest1.align.secY.codon.50.label.fasta = A FASTA file containing multiple alignment of a single gene (secY), codon aligned, 50% filtered with full label
- mytest1.align.secY.codon.50.zZ.fasta = A FASTA file containing multiple alignment of a single gene (secY), codon aligned, 50% filtered with zZ+Unique id+zZ
- finally3.UBCG_gsi(92).codon.50.label.nwk
Test run using example data
An example set is provided with the UBCG package. Please follow the instruction below:
- Unzip the UBCG package. Example fasta files should be in “fasta” directory. Execute the commands given here to convert fasta files to bcg files.
- Check the “bcg” directory by “ls bcg/*” where you should be able to find eight *.bcg files that contain UBCG gene sequences with metadata.
- To align and generate the UBCG tree, execute the following line
- java -jar UBCG.jar align -bcg_dir bcg -prefix my_example
- Outputs will be saved in “output/my_example” directory
- Take the “*.UBCG_gsi(92).codon.50.label.nwk” file and open with MEGA or other tree-viewing programs. (The below is the screenshot of MEGA.)
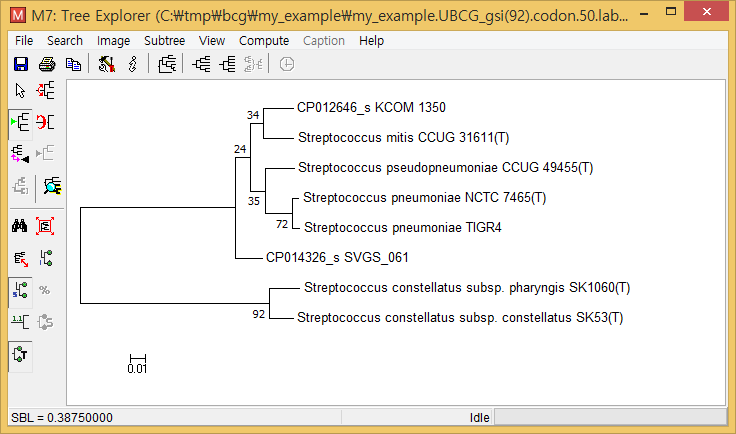
This UBCG tree shows that
- Two Streptococcus constellatus subspecies are closely related; all 92 UBCGs supported this.
- Streptococcus pneumoniae TIGR4 is closely related to the type strain of Streptococcus pneumoniae; 72 out of UBCGs supported this.
- Two tentatively new species, named CP012646_s and CP014326_s, formed a monophyletic clade with S. pneumoniae, Streptococcus pseudopneumoniae, and Streptococcus mitis.
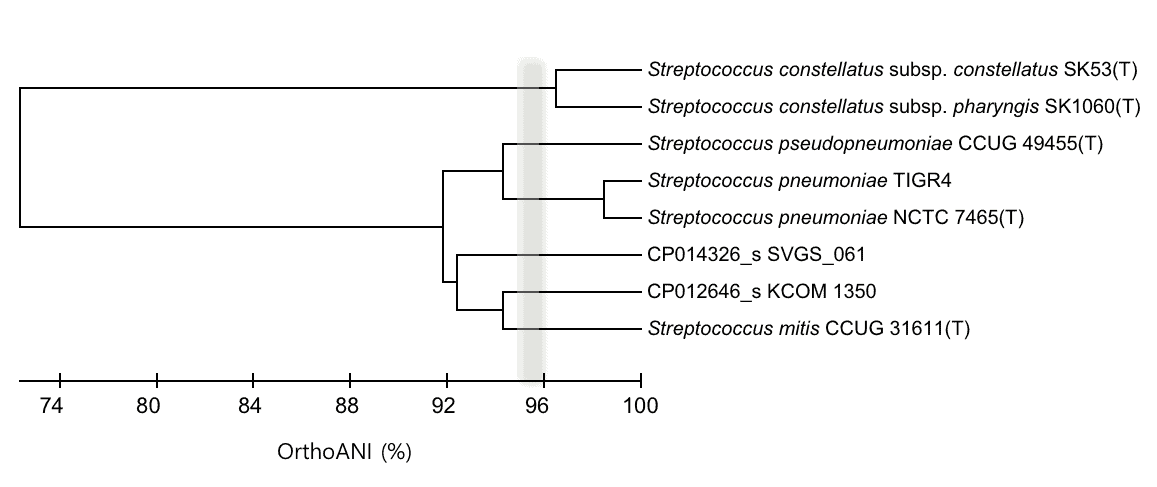
The below is the dendrogram showing OrthoANI-based clustering of the same genomes. Please note that CP012646_s and CP014326_s represent a novel species using 95~96% average nucleotide identity (ANI) cutoff [Learn more].
Frequently asked questions
- How can I access the UBCG sequences?
- Use our UBCG viewer at https://www.ezbiocloud.net/tools/ubcg_viewer. to open any *.bcg file. Click the right mouse button to copy DNA or protein sequences of any UBCG into the clipboard.
Last updated March 1, 2018, by JC

