We will work through with an example file. Please download “Leuconostoc_16s.ezb” here and open it with EzEditor2.
This file contains 16S sequences of the type strains of the genus Leuconostoc. By opening it, you will see the “Select Window” showing metadata of 19 sequences.
Click icon (1) to start “Align Window”.
Now, we are in the “Align Window”. Here, we can browse alignment and edit multiple alignment. In 16S mode, secondary structure information is displayed to help your manual adjustment. Click here to see typical secondary structures of bacterial 16S rRNA.
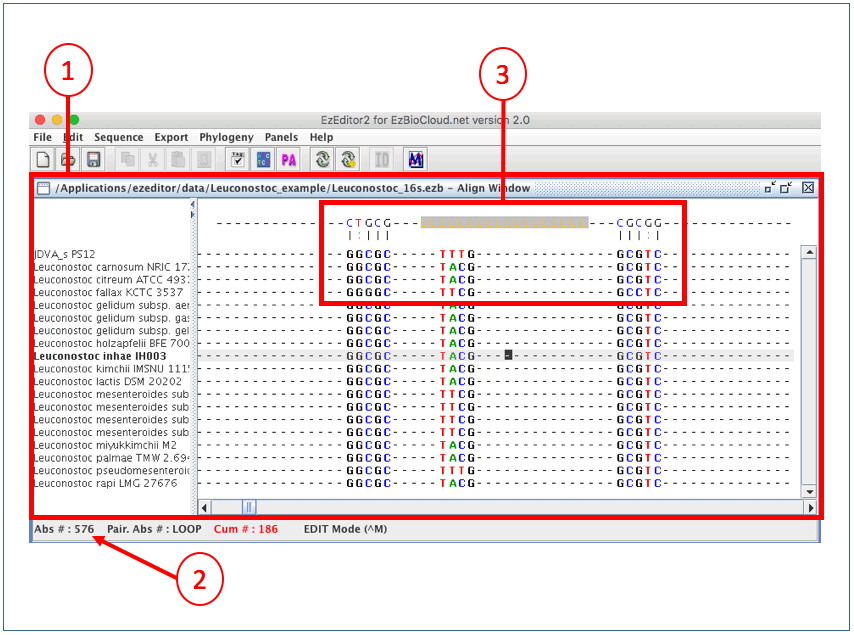
(1) This is the “Align Window”.
(2) Move the cursor to around absolute position of #567.
(3) This is an area displaying secondary structure.
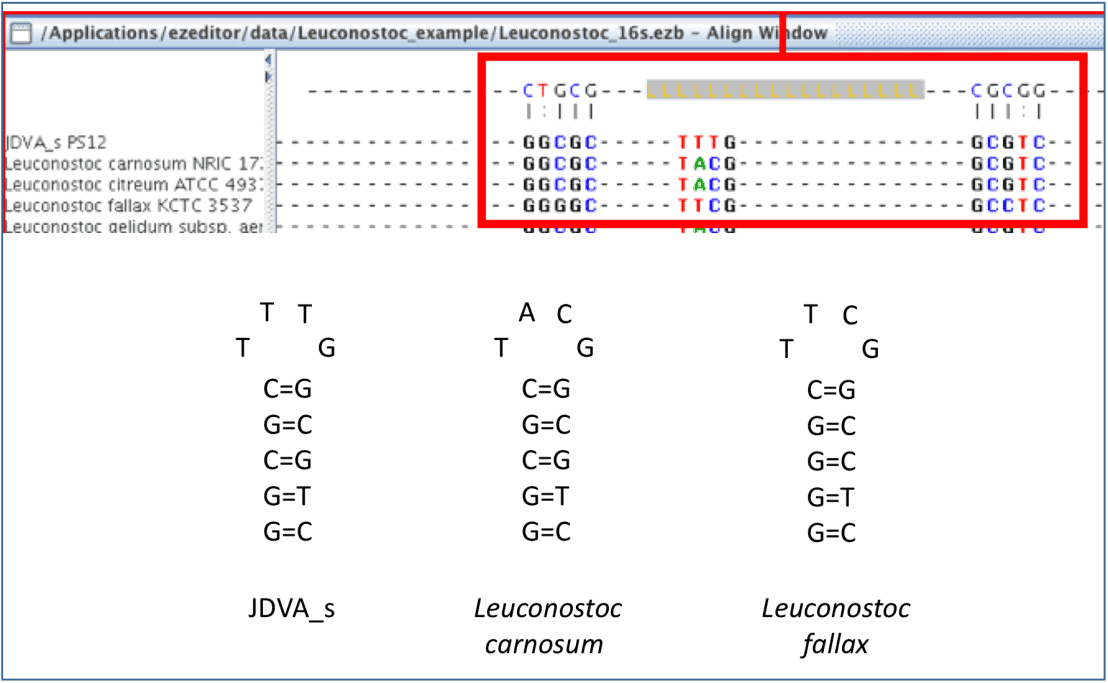
The way that EzEditor shows the secondary structure of 16S rRNA gene is intuitive. The bases of positions where hydrogen bonds occur are displayed in the panel at the top of the Select Window. We will show you the exact secondary structures that are represented in EzEditor’s screen, below. Anyone can easily find odd or erroneous bases that do not form a secondary structure where they should do. Please note that [LLL..LLL] in the top panel indicates the loop region of a hairpin structure.
Adding new sequence
The following is a sequence we will add to the EzEditor file. Copy this FASTA format into clipboard and paste into the current EzEditor “Select Window” by Control+V key
>My query GCTGGCGGCGTGCCTAATACATGCAAGTCGAACGCACAGCGAAAGGTGCTTGCACCTTTCAAGTGAGTGGCGAACGGGTGAGTAACACGTGGATAACCTACCTCAAGGTTGGGGATAACATTTGGAAACAGATGCTAATACCGAATAAAACTTCGTATCGCATGATACAAGGTTAAAAGGCGCTACGGCGTCACCTAGAGATGGATCCGCGGTGCATTAGTTAGTTGGTGGGGTAAAGGCCTACCAAGACAATGATGCATAGCCGAGTTGAGAGACTGATCGGCCACATTGGGACTGAGACACGGCCCAAACTCCTACGGGAGGCTGCAGTAGGGAATCTTCCACAATGGGCGAAAGCCTGATGGAGCAACGCCGCGTGTGTGATGAAGGCTTTCGGGTCGTAAAGCACTGTTGTATGGGAAGAACAGCTAGAGTAGGGAGTGACTTTAGTTTGACGGTACCATACCAGAAAGGGACGGCTAAATACGTGCCAGCAGCCGCGGTAGTACGTATGTCCCGAGCGTTATCCGGATTTATTGGGCGTAAAGCGAGCGCAGACGGTTGATTAAGTCTGATGTGAAAGCCCGGAGCTCAACTCCGGAAAGGCATTGGAAACTGGTCAACTTGAGTGCAGTAGAGGTAAGTGGAACTCCATGTGTAGCGGTGGAATGCGTAGATATATGGAAGCACACCAGCGGCGAAGGCGCTTACTGGACTGTAACTGACGTTGAGGCTCGAAAGTGTGGGTAGCAAACAGGATTAGATACCCTGGTAGTCCACACCGTAAACGATGAACACTAGGTGTTAGGAGGTTTCCGCCTCTTAGTGCCGAAGCTAACGCATTAAGTGTTCCGCCTGGGGAGTACGACCGCAAGGTTGAAACTCAAAGGAATTGACGGGGACCCGCACAAGCGGTGGAGCATGTGGTTTAATTCGAAGCAACGCGAAGAACCTTACCAGGTCTTGACATCCTTTGAAGCTTTTAGAGATAGAAGTGTTCTCTTCGGAGACAAAGTGACAGGTGGTGCATGGTCGTCGTCAGCTCGTGTCGTGAGATGTTGGGTTAAGTCCCGCAACGAGCGCAACCCTTATTGTTAGTTGCCAGCATTCAGATGGGCACTCTAGCGAGACTGCCGGTGACAAACCGGAGGAAGGCGGGGACGACGTCAGATCATCATGCCCCTTATGACCTGGGCTACACACGTGCTACAATGGCGTATACAACGAGTTGCCAACCTGTGAAGGTGAGCTAATCTCTTAAAGTACGTCTCAGTTCGGACTGCAATCTGCAACTCGACTGCACGAAGTCGGAATCGCTAGTAATCGCGGATCAGCACGCCGCGGTGAATACG
After importing a sequence, you should see it in both “Select Window” and “Align Window”
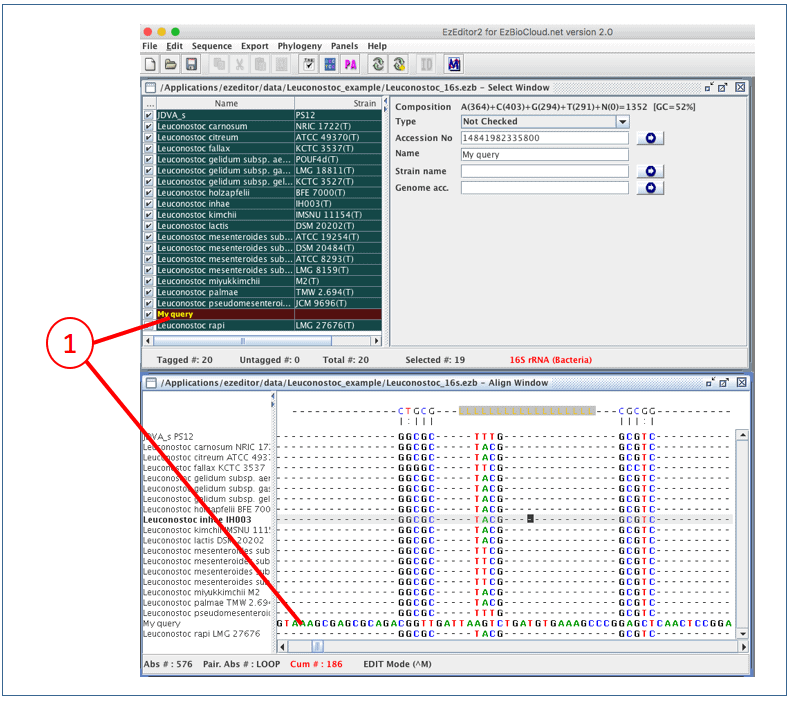
(1). Please note that your sequence is not yet aligned.
To align this new sequence to the existing aligned sequences, we want to place our newly imported sequence at the bottom of “Align Window”. Currently, “Leuconostoc rapi” is displayed at the bottom. Simply, unselect “Leuconostoc rapi” in Select Window, so it will disappear in Align Window.

Now, we are ready for semi-automated alignment.
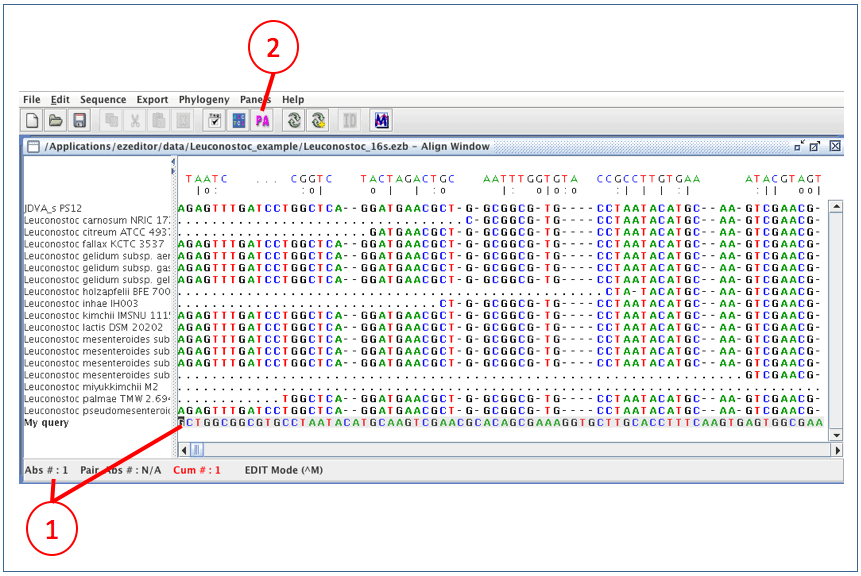
(1) Move the window and cursor to the most left-bottom position. It is important to place the cursor on our new sequence.
(2) Click [PA] for pairwise alignment. This will invoke an aligner which will align your sequence to a template sequence of your choice.
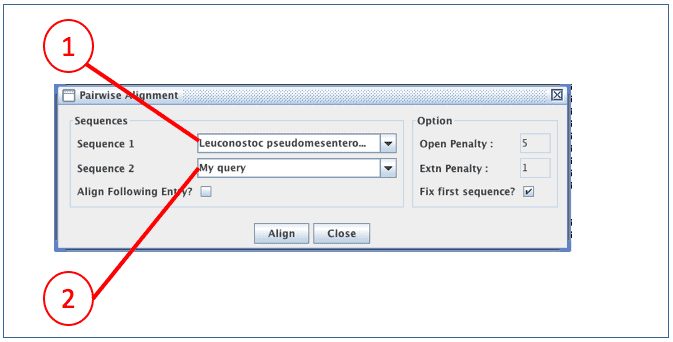
(1) The sequence that will be used as a template for alignment. Alignment of this sequence is not changed. It is best to select the sequence which is most similar to the sequence to be aligned.
(2) This is the sequence that you are going to align. EzEditor will try to align at its best, but you may need to adjust manually later.
Now new sequence is aligned (as you will see right away).
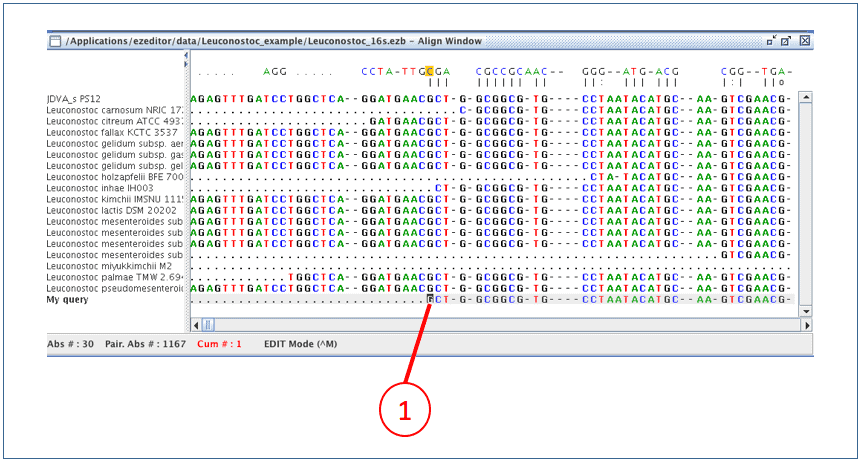
(1) Move the cursor to the starting position of the sequence.
Now on, we will inspect the alignment and adjust as needed. Click the right mouse button to see the possible keys to use. We recommend using Control+U key which will move the cursor quickly to the position that does not match to the sequence at the right above. We do not need to check the regions manually if the new sequence is same as the others.
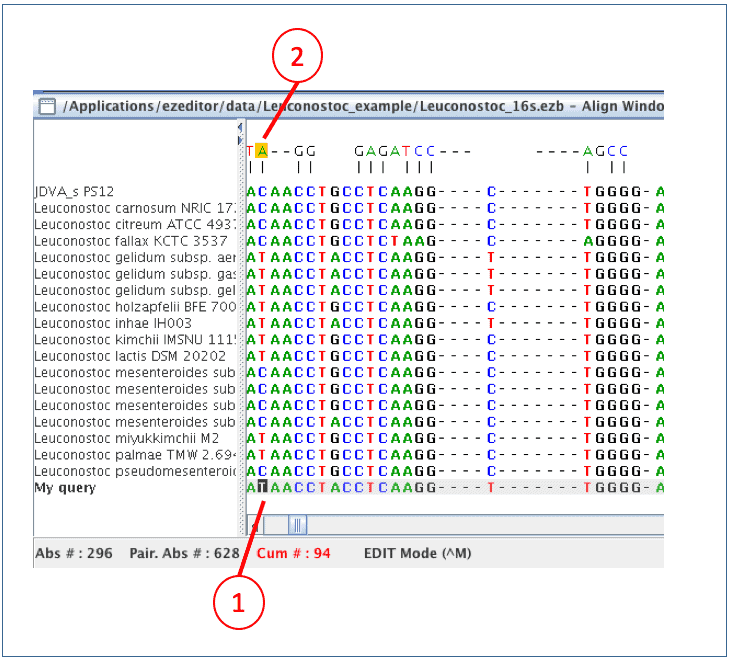
(1) The cursor is now at the position that is different from one at the above. It is [T] whereas the above (=Leuconostoc pseudomesenteroides) has [C].
(2) [T] formed a pair with [A], so it seems a good sequence (not from sequencing errors)
You will look for the next position with a discrepancy by the Control+U key. Adjust alignment by adding gap (space key) or deleting gap (DEL key). In this example, EzEditor does an almost perfect job so you would not probably need manual adjustments. However, in many cases, manual alignment requires substantial experience. We hope that secondary structure information can help you with this task to save time and improve accuracy. Please note that all 16S sequences in EzBioCloud.net are manually aligned using this tool to maximize the accuracy of subsequent analyses.
Go to “phylogenetic analysis using EzEditor2”
Go back to the content of tutorials
Last modified on January 12, 2017 (JC)

