Microbiome bioinformatics
Microbiome
A microbiota is the entire collection of microorganisms in a specific niche, such as the human gut or soil. The microbiome is comprised of all of the genetic material within a microbiota. The most important methodology for studying microbiomes is metagenomics which involves the massive sequencing of DNA followed by sophisticated bioinformatics.
The goals of microbiome research are to understand (i) who are the inhabitants (ii) what they do and (iii) how they do it.
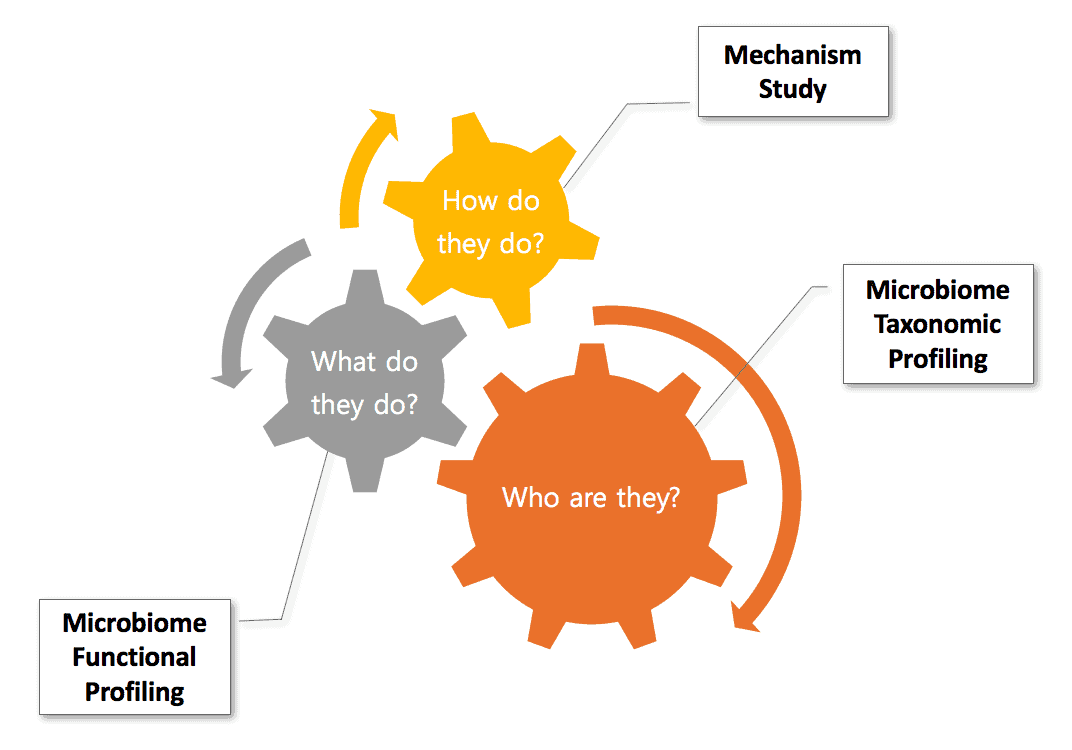
Goals of the microbiome study
To achieve these goals, we need to get the taxonomic and functional profiles of microbiome samples, then group and compare them to understand differences. For example, we could identify bacterial species responsible for causing obesity by comparing taxonomic profiles between the groups of healthy and obese subjects. High throughput DNA sequencing provides an accurate and efficient way to obtain these profiles.
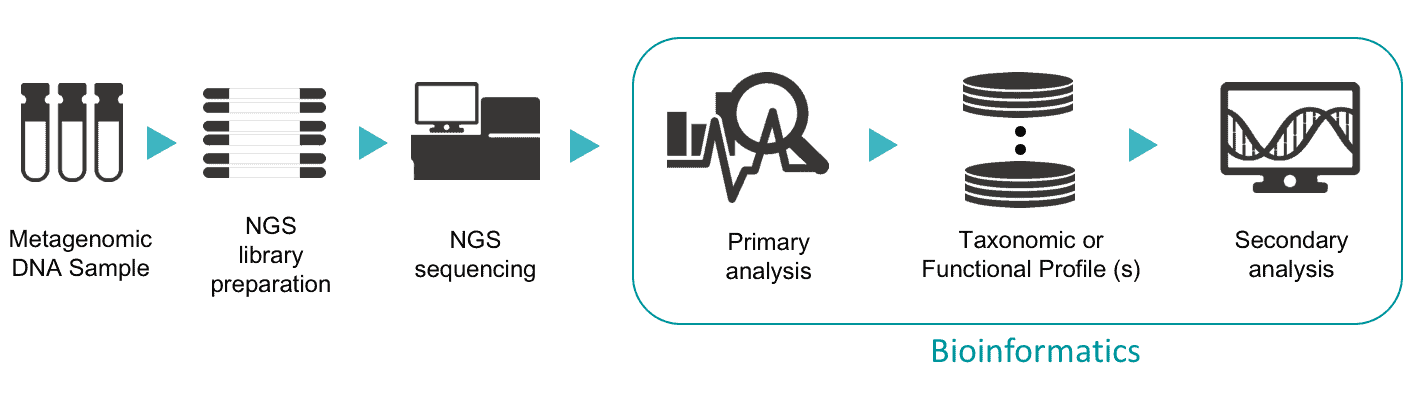
Steps in Microbiome Bioinformatics
The above figure summarizes the major steps in microbiome studies. The process of bioinformatics can be divided into two steps: primary and secondary analyses.
Primary analysis in microbiome bioinformatics
In this step, NGS reads in large volume are turned into light-weight profiles. For example, if 100,000 16S NGS reads match to the sequence of Vibrio cholerae type strain, the final profile will only store only the count information, i.e., 100,000, of V. cholerae, not the raw sequence data. Similarly, NGS sequences matched to a certain functional ortholog group, e.g., K00076 involved in secondary bile acids biosynthesis, will be stored in the functional profile with only the count information. A series of software tools, called a pipeline, is used to process raw NGS data in order to generate taxonomic or functional profiles.
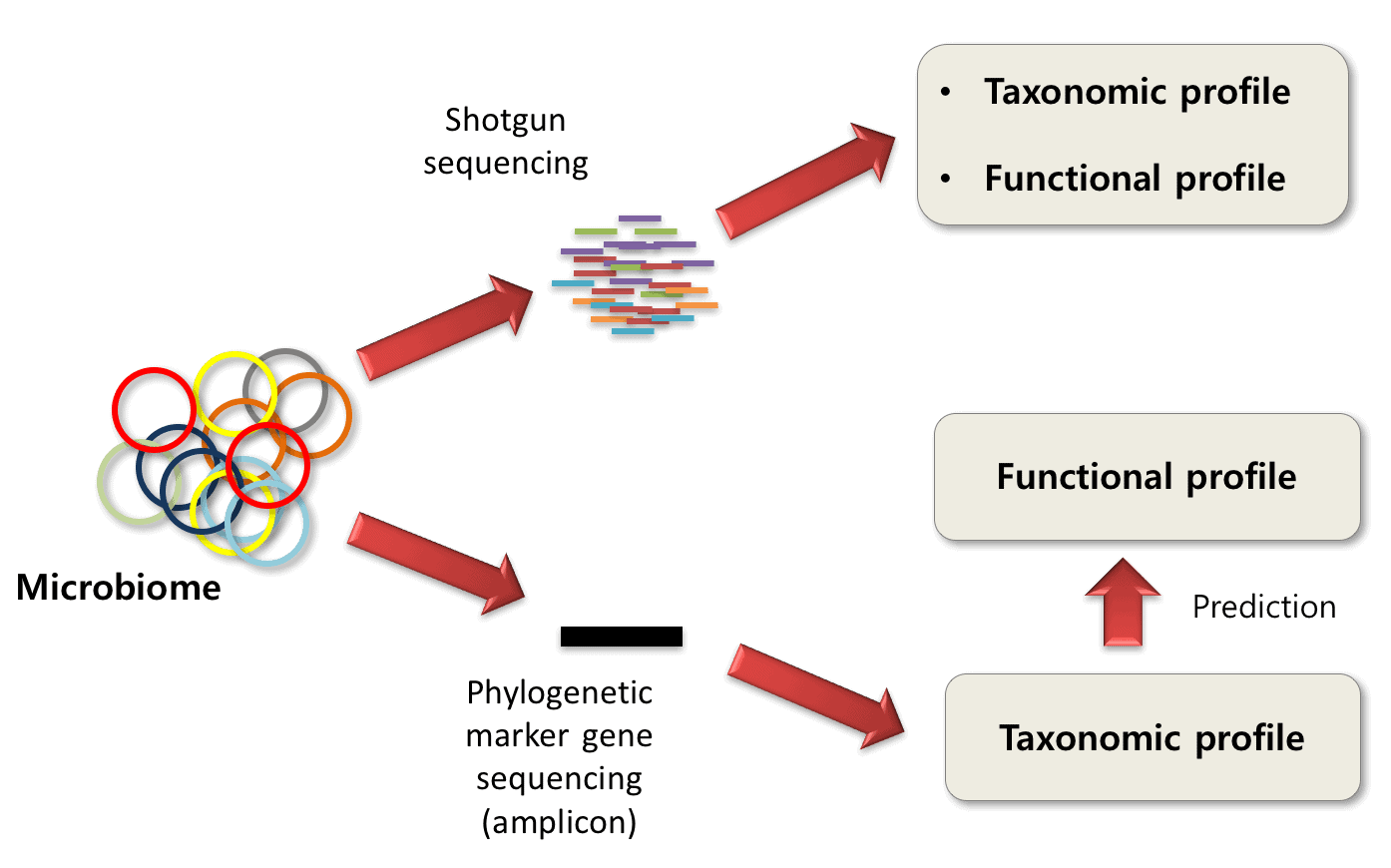
Workflow in metagenomics
The most popular method of generating microbiome profiles is by sequencing amplicons of phylogenetic markers. 16S and ITS are the choice of markers genes for Bacteria and fungi, respectively. It is both cheap and sufficient to capture the taxonomic structure of microbiome samples. The drawback is that only taxonomic profiles can be obtained. To obtain functional profiles, shotgun sequencing should be used. There is a way of predicting functional profiles from taxonomic profiles (See Langille et al., 2013), but the accuracy cannot be guaranteed. The following table illustrates the pros and cons of amplicon and shotgun metagenomics.
16S copy number correction
What is the 16S copy number and why it matters?
The 16S rRNA gene (16S) has been widely used as a phylogenetic marker, particularly important for the taxonomic profiling of microbiome samples. Unlike other genes that code for proteins, the 16S-coding gene may be present in multiple copies in a single cell. Obviously, a bacterial strain must have at least one gene encoding 16S, but the copy number can go up to 15 (see the below chart). There is a positive correlation between the genome size and 16S copy number.

16S copy numbers of bacteria in EzBioCloud database (generated from only complete genomes).
When we analyze microbiome data using 16S amplicon sequences, all quantitative measures are a form of NGS read counts that are assigned to the known taxa. In this case, we actually count the number of a marker gene, typically 16S, present in a microbiome sample. However, what we eventually want to know is not the number of 16S reads but the number of corresponding cells, or CFU (colony forming units).
Let’s assume that we are analyzing a human fecal sample. After sequencing, we obtained 100 reads assigned to Bacteroides fragilis and also 100 to Prevotella copri. Both species are frequently found in the human gut. Should we say that two species are present in equal numbers? According to the EzBioCloud database which provides information about 16S copy number, B. fragilis has 6 copies whereas P. copri contains 4 copies [Learn more for B. fragilis and P. copri]. If we consider this, the corrected ratio between B. fragilis and P. copri should be 3:2, not 1:1. The necessity of 16S copy number correction or normalization has been raised by several studies (Kembel et al., 2012; Angly et al., 2014; Vandeputte, et al. 2017).
How to correct taxonomic profile data using 16S copy numbers
The relative taxonomic compositional data of a microbiome sample can be corrected by simple calculation once we know the 16S copy numbers of all species. A problem is that we do not know these values for all species. To obtain accurate data, one or more complete genome sequence is required. Incomplete genome assemblies derived from short NGS reads contain either no or an inaccurate number of 16S gene sequences. At present, there are 3467 species represented by complete genome sequences (As of Dec. 2017). 16S copy numbers of the remaining species, including uncultured phylotypes, should be interpolated using the existing data.
A couple of algorithms were proposed to predict the missing 16S copy numbers (Langille et al., 2013; Angly et al., 2014). In EzBioCloud 16S-based MTP app, the PICRUSt algorithm (Langille et al., 2013) is used to generate the 16S copy number database for all species/phylotypes in the EzBioCloud 16S database (the below figure).
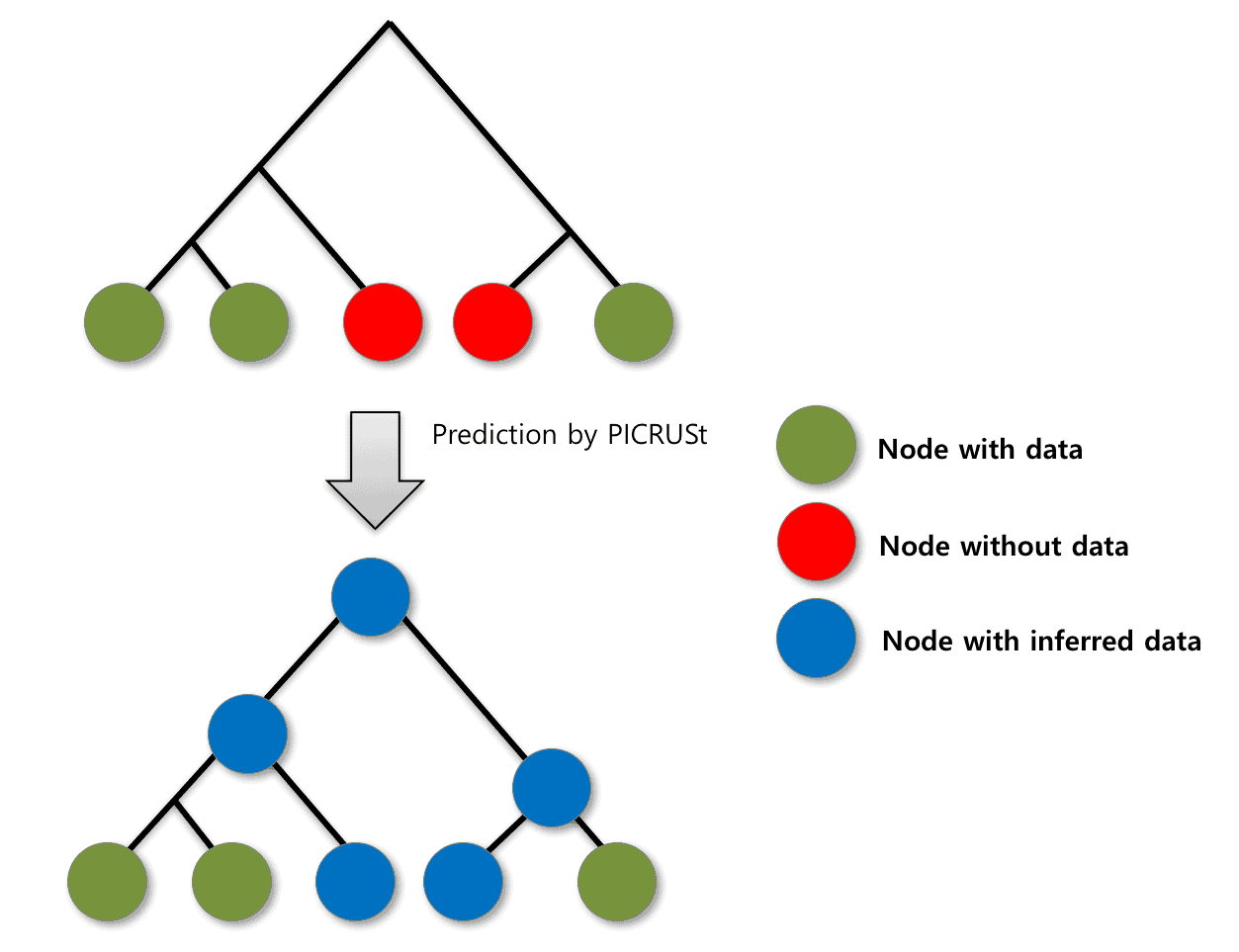
Prediction of 16S copy numbers using PICRUSt algorithm
Implementation in EzBioCloud 16S-based MTP app
EzBioCloud 16S-based MTP app allows you to instantly and interactively apply 16S copy number correction to the comparative analysis of multiple samples, the calculation of beta-diversity, and to Biomarker Discovery (e.g., LefSe) as well. Our database of 16S copy number is more comprehensive than any other database as we utilize an up-to-date version of the genome database (8,631 quality-controlled genomes of 3,302 species; as of March 2018).
The EzBioCloud team / Last edited on Mar. 6, 2018

