This document explains how to use EzBioCloud’s “Identity” service using single Sanger sequencing data.
Preparations
- Use ab1 file but not a text file. An ab1 file contains the chromatogram information from ABI sequencers (Learn more). If your sequencing service provider did not give you ab1 files, ask for ab1 files.
- Install the ab1 file viewer. There are several free software tools.
- For Mac OS X, use 4Peaks tool [Download].
- Fox MS Windows, use chromas tools [Download].
- You need a sequence alignment tool for 16S gene sequences. We recommend the EzEditor2 tool [Download] [Tutorial of EzEditor2]. The following example ab1 files are required.
Characteristics of Sanger sequencing data
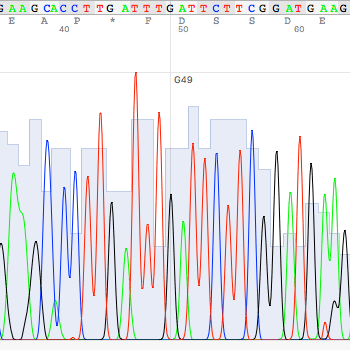
- An ab1 file contains both the chromatogram and sequence data (after base calling). Quality and manual inspection of the raw data is possible with the chromatogram. Because of the sequencing chemistry, both ends of sequence contain substantial errors. If you used the sequence data without trimming the ends, your sequence will have a substantial amount of errors.
A Case of good quality
- Download case #1 ab1 file here. This contains a 16S sequence of a human fecal bacterium.
- Open chromatogram using a viewer. I used 4Peaks tool. This file contains the result of the fairly good sequencing reaction.

- Copy sequence into the clipboard. Then, search through the EzBioCloud’s Identify. Go to https://www.ezbiocloud.net/identify and paste your sequence as new query. The result will be like this:
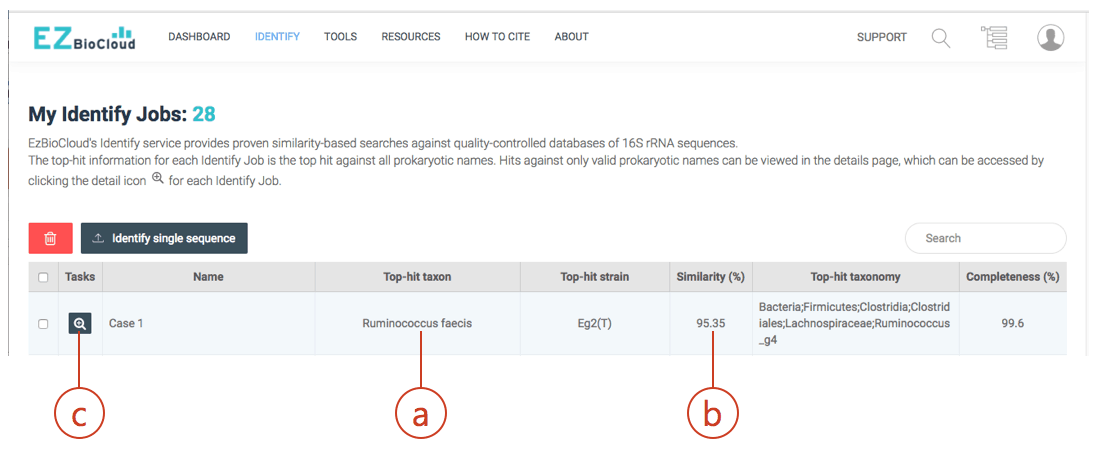
The hit species (a) is displayed with pairwise sequence similarity (b). In this case, sequence similarity is 95.35% and you may be excited to discover a potentially new species. The generally accepted 16S similarity cutoff is 98.7% [Learn more]. This is cutoff is applicable for high-quality sequence only. As you will see the chromatogram shown above, your sequence contains errors at the front end (as well as backend). We will start the trimming process using a sequence alignment tool called EzEditor2. Click (c) to view the detailed result of “Identify”.

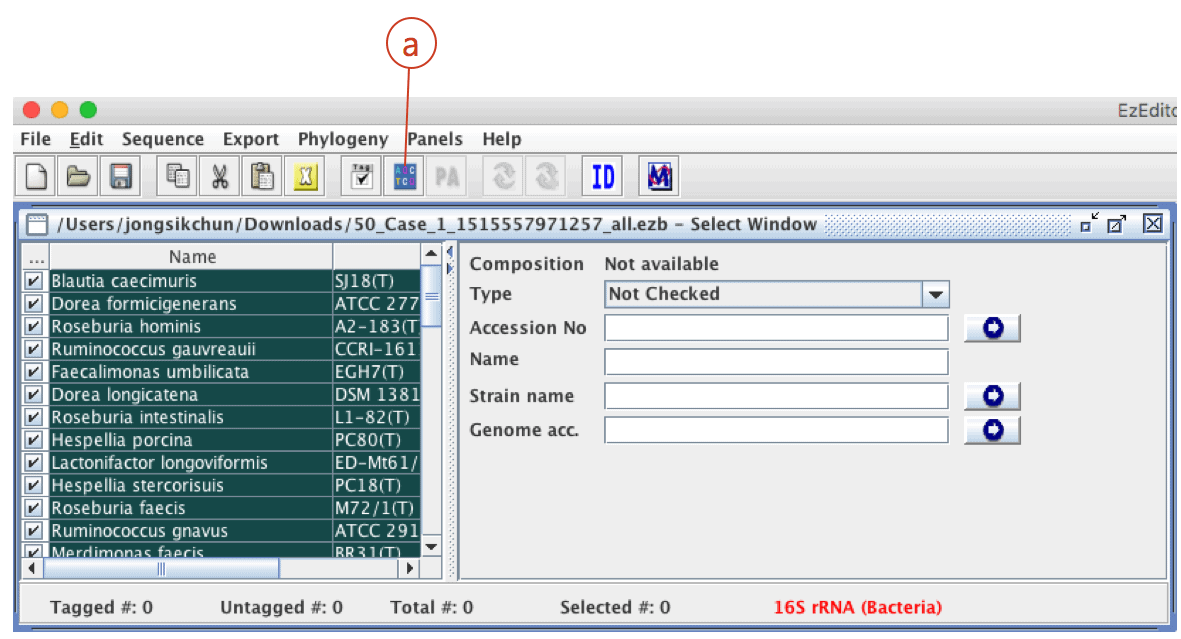
Click (a) to download a data file for EzEditor2 tool. This file contains your sequences and reference sequences that show the highest sequence similarities.
Start EzEditor2 software and open the file that you have just downloaded. You will see the alignment like this:
Click (a) to open “alignment panel”.
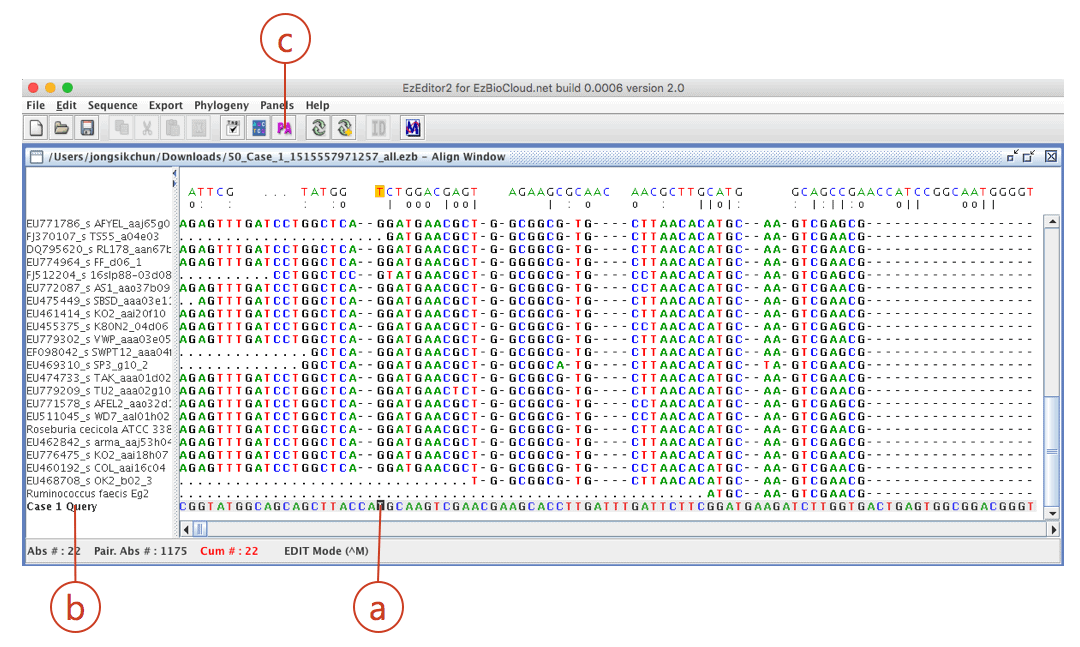 Your sequence is located at the bottom of the alignment. Move the cursor (a) to the bottom of the screen. Check your sequence (from ab1 file) is at the bottom (b). The sequence of the best hit (Ruminococcus faecis) is located at the right upper row of your sequence).
Your sequence is located at the bottom of the alignment. Move the cursor (a) to the bottom of the screen. Check your sequence (from ab1 file) is at the bottom (b). The sequence of the best hit (Ruminococcus faecis) is located at the right upper row of your sequence).
Alignment your sequence against the hit sequence (Ruminococcus faecis) by clicking [PA](c).
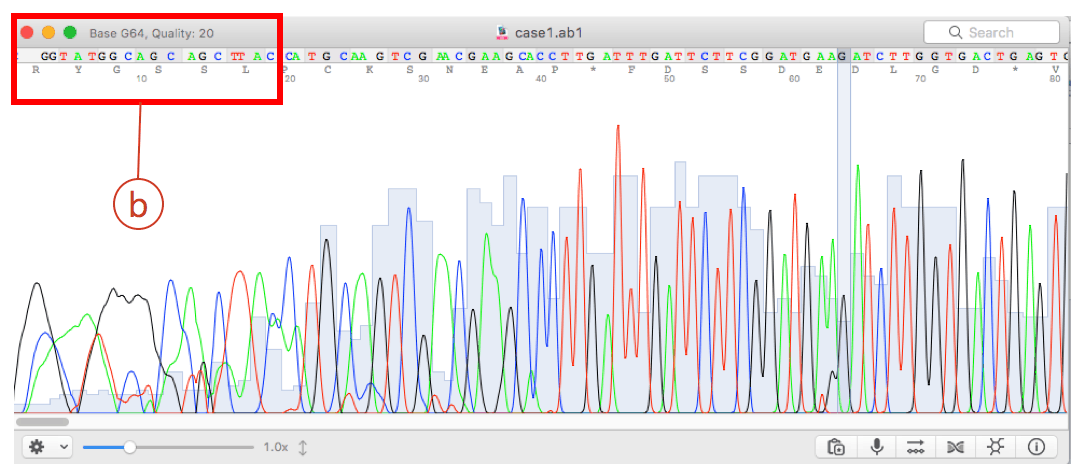
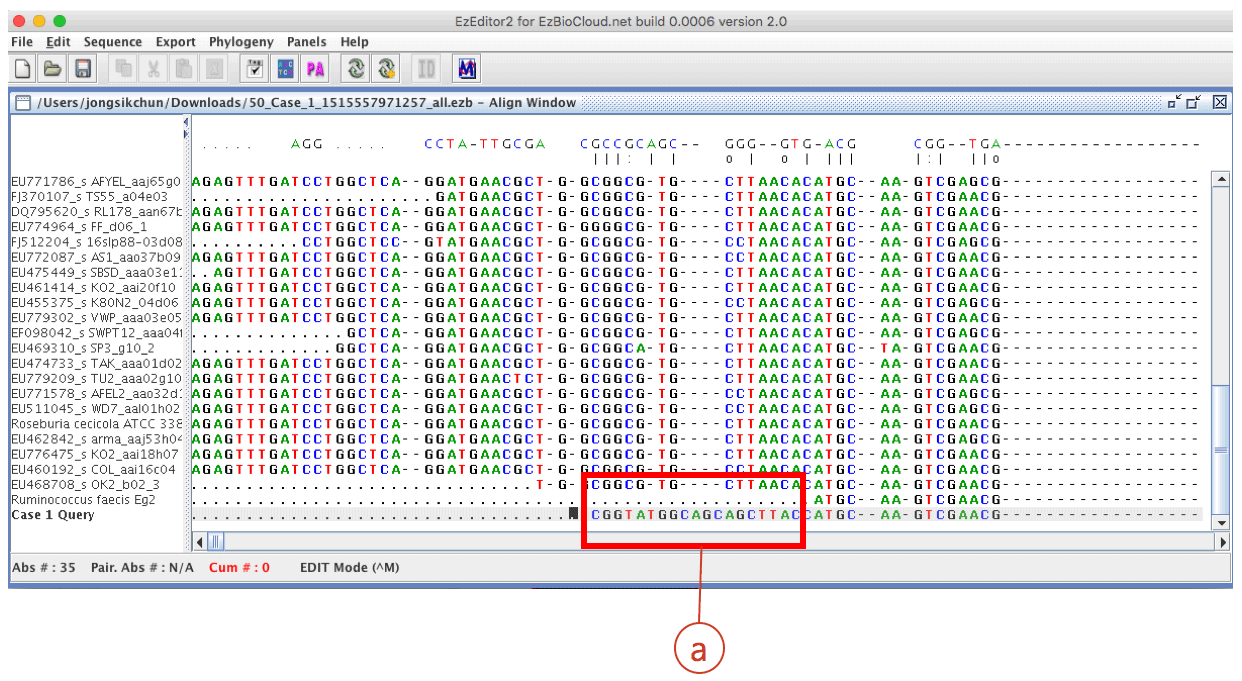 After the automated pairwise alignment, you will see the erroneous region that does not match to other reference sequences (box (a) of the above). The corresponding region in the chromatogram also shows low quality sequencing reaction (box (b) of the blow). Both boxes depict the same region.
After the automated pairwise alignment, you will see the erroneous region that does not match to other reference sequences (box (a) of the above). The corresponding region in the chromatogram also shows low quality sequencing reaction (box (b) of the blow). Both boxes depict the same region.
You need to trim this erroneous region in the EzEditor2 tool. Move the cursor to the position where you want to trim. Right-click to activate the popup menu then select “Delete”->”Before cursor”. Or use the shortcut key (Shift+A). See (a) below.
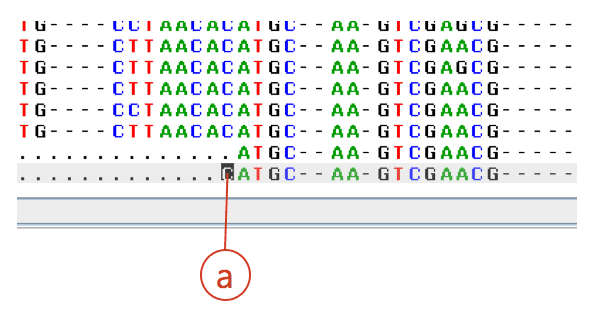
You will do the same for the backend. A tip is to use the shortcut Ctrl+U key (Compare UP function). This will move the cursor to the next position where nucleotide does not match to the hit reference sequence. Compare your sequence with the hit reference sequence and also consult the chromatogram. Find the position where you want to trim the right-side (the rest of sequence).
You must be very conservative. 400 bp is sufficient to get an accurate identification. Trim as much as possible. In this example, the ab1 contains 1,539 bases (very long for a single Sanger reaction). I trimmed at the absolute position of 1,213 which left 519 bases. The trimmed sequence can be obtained here as FASTA, EzEditor2 file.
 To copy your trimmed sequence, (a) select “Select Panel” and (b) select “Copy as FASTA format”. Paste to the EzBioCloud “Identify” as a new query. The result is shown below:
To copy your trimmed sequence, (a) select “Select Panel” and (b) select “Copy as FASTA format”. Paste to the EzBioCloud “Identify” as a new query. The result is shown below:
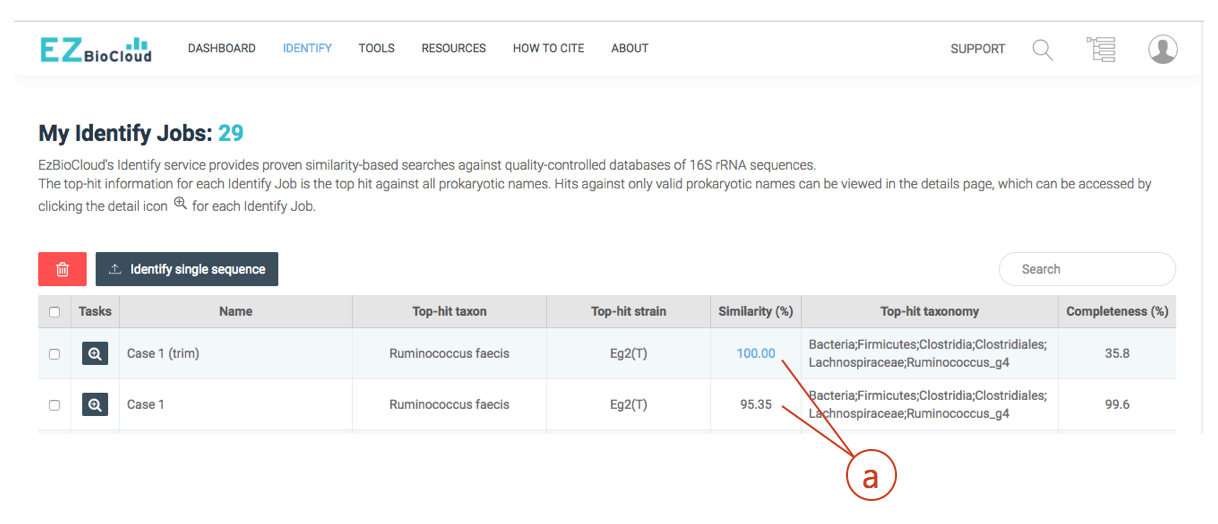 (a) After trimming, your sequence is 100% identical to the hit (Ruminococcus faecis). Even though there are cases where the different species show identical 16S sequences, this case clearly indicates that the isolate belongs to Ruminococcus faecis.
(a) After trimming, your sequence is 100% identical to the hit (Ruminococcus faecis). Even though there are cases where the different species show identical 16S sequences, this case clearly indicates that the isolate belongs to Ruminococcus faecis.
A Case of bad quality
Any region of low-quality chromatogram should be trimmed. In the worst cases, you will need to sequence again. The example below looks OK at a first glance, but this region should be trimmed.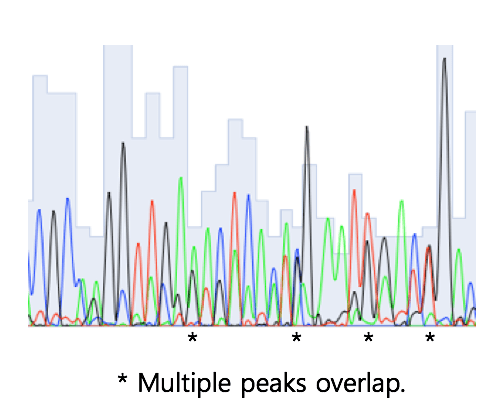
Conclusions
- Use only a good-quality region of your sequence.
- Select the good-quality region using multiple alignments with high-quality reference sequences and manual inspection of the chromatogram.
Upadted by Jon Jongsik Chun on Jan 10, 2018.

